Prof. Dr. Serena DeBeer - Inorganic Spectroscopy

- Prof. Dr. Serena DeBeer
- Director
- Inorganic Spectroscopy
- +49 (0)208 306 - 3656
- serena.debeer(at)cec.mpg.de
- Room: 608
Vita
| B.S. (Chemistry) | Southwestern University, TX, USA (1995) |
| Ph.D. (Chemistry) | Stanford University, CA, USA (2002) |
| Beam Line Scientist | SSRL, SLAC, Stanford, University (2001-2003) |
| Staff Scientist | SSRL, SLAC, Stanford, University (2003-2009) |
| Assistant Professor | Cornell University, NY, USA (2009-2012) |
| Research Group Leader | MPI for Bioinorganic Chemistry; today: MPI CEC (2011-2017) |
| Group leader | PINK Beamline, Energy Materials In-Situ Laboratory, Helmholtz Zentrum, Berlin (since 2012) |
| Adjunct Associate Professor | Department of Chemistry and Chemical Biology, Cornell University (since 2012) |
| Honorary Faculty | Ruhr University Bochum (since 2014) |
| Director | Inorganic Spectroscopy, MPI CEC (since 2017) |
| Honorary Faculty | University of Duisburg-Essen (since 2024) |
Publications
Full publications list | ORCID | ResearcherID | Google Scholar Profile
MPI CEC publications
- Chrysina, M., Drosou, M., Castillo, R. G., Reus, M., Neese, F., Krewald, V., Pantazis, D. A., DeBeer, S. (2023). Nature of S-States in the Oxygen-Evolving Complex Resolved by High-Energy Resolution Fluorescence Detected X-ray Absorption Spectroscopy. Journal of the American Chemical Society, 145(47), 25579-25594. doi:10.1021/jacs.3c06046.
- Sodreau, A., Zahedi, H. G., Dervisoglu, R., Kang, L., Menten, J., Zenner, J., Terefenko, N., DeBeer, S., Wiegand, T., Bordet, A., Leitner, W. (2023). A Simple and Versatile Approach for the Low-Temperature Synthesis of Transition Metal Phosphide Nanoparticles from Metal Chloride Complexes and P(SiMe3)3. ADVANCED MATERIALS. 2306621 (1-9) doi:10.1002/adma.202306621.
- Wandzilak, A., Grubel, K., Skubi, K. L., McWilliams, S. F., Bessas, D., Rana, A., Hugenbruch, S., Dey, A., Holland, P. L., DeBeer, S. (2023). Mössbauer and Nuclear Resonance Vibrational Spectroscopy Studies of Iron Species Involved in N-N Bond Cleavage. INORGANIC CHEMISTRY, 62(45), 18449-18464. doi:10.1021/acs.inorgchem.3c02594.
- DeBeer, S., Moonshiram, D. (2023). Mapping the Ultrafast Mechanistic Pathways of Co Photocatalysts in Pure Water through Time-Resolved X-ray Spectroscopy. ChemSusChem, e202300719, pp. 1-14. doi:10.1002/cssc.202300719.
- Pang, Y., Nöthling, N., Leutzsch, M., Kang, L., Bill, E., van Gastel, M., Reijerse, E., Goddard, R., Wagner, L., SantaLucia, D., DeBeer, S., Neese, F., Cornella, J. (2023). Synthesis and isolation of a triplet bismuthinidene with a quenched magnetic response. Science, 380 (6649), 1043-1048. doi:10.1126/science.adg2833.
- Genoux, A., Pauly, M., Rooney, C. L., Choi, C., Shang, B., McGuigan, S., Fataftah, M. S., Kayser, Y., Suhr, S. C. B., DeBeer, S., Wang, H., Maggard, P. A., Holland, P. L. (2023). Well-Defined Iron Sites in Crystalline Carbon Nitride. JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 145(38), 20739-20744. doi:10.1021/jacs.3c05417.
- Zhang, Y., El Sayed, S., Kang, L., Sanger, M., Wiegand, T., Jessop, P. G., DeBeer, S., Bordet, A., Leitner, W. (2023). Adaptive Catalysts for the Selective Hydrogenation of Bicyclic Heteroaromatics using Ruthenium Nanoparticles on a CO2-Responsive Support. Angewandte Chemie, International Edition in English, (XX): e202311427, pp. x-xx. doi:10.1002/anie.202311427.
- Hau, J.-L., Kaltwasser, S., Muras, V., Casutt, M. S., Vohl, G., Claussen, B., Steffen, W., Leitner, A., Bill, E., Cutsail, G. E., DeBeer, S., Vonck, J., Steuber, J., Fritz, G. (2023). Conformational coupling of redox-driven Na+-translocation in <i>Vibrio cholerae</i> NADH:quinone oxidoreductase. Nature Structural & Molecular Biology, (xx), 1-31. doi:10.1038/s41594-023-01099-0.
- Erbe, A., Tesch, M. F., Rüdiger, O., Kaiser, B., DeBeer, S., Rabe, M. (2023). Operando studies of Mn oxide based electrocatalysts for the oxygen evolution reaction. Physical Chemistry Chemical Physics, (40), 26933-27894. doi:10.1039/d3cp02384b.
- Liu, Y., Chatterjee, S., Cutsail III, G. E., Peredkov, S., Gupta, S. K., Dechert, S., DeBeer, S., Meyer, F. (2023). Cu4S Cluster in "0-Hole" and "1-Hole" States: Geometric and Electronic Structure Variations for the Active Cu-Z* Site of N2O Reductase. Journal of the American Chemical Society, 145(33), 18477-18486. doi:10.1021/jacs.3c04893.
- Hall, K. R., Joseph, C., Ayuso-Fernandez, I., Tamhankar, A. V., Rieder, L., Skaali, R., Golten, O., Neese, F., Rohr, A. K., Venturinelli Jannuzzi, S. A., DeBeer, S., Eijsink, V. G. H., Sorlie, M. (2023). A Conserved Second Sphere Residue Tunes Copper Site Reactivity in Lytic Polysaccharide Monooxygenases. Journal of the American Chemical Society, 145(34), 18888-18903. doi:10.1021/jacs.3c05342.
- Mao, W., Zhang, Z., Fehn, D., Venturinelli Jannuzzi, S. A., Heinemann, F., Scheurer, A., van Gastel, M., DeBeer, S., Munz, D., Meyer, K. (2023). Synthesis and Reactivity of a Cobalt-Supported Singlet Nitrene. Journal of the American Chemical Society, 145(25), 13650-13662. doi:10.1021/jacs.3c01478.
- Pielsticker, L., Nicholls, R. L., DeBeer, S., Greiner, M. (2023). Convolutional neural network framework for the automated analysis of transition metal X-ray photoelectron spectra. Analytica Chimica Acta, (1271) 341433 doi:10.1016/j.aca.2023.341433.
- Van Stappen, C., Benediktsson, B., Rana, A., Chumakov, A., Yoda, Y., Bessas, D., Decamps, L. B., Bjornsson, R., DeBeer, S. (2023). Structural correlations of nitrogenase active sites using nuclear resonance vibrational spectroscopy and QM/MM calculations. Faraday Discussions, (243) 253-269.. doi:10.1039/d2fd00174h.
- Kovel, C. B., Darmon, J. M., Stieber, S. C. E., Pombar, G., Pabst, T. P., Theis, B., Turner, Z. R., Ungör, Ö., Shatruk, M., DeBeer, S., Chirik, P. J. (2023). Bimolecular Reductive Elimination of Ethane from Pyridine(diimine) Iron Methyl Complexes: Mechanism, Electronic Structure, and Entry into [2+2] Cycloaddition Catalysis. Journal of the American Chemical Society, (145), 5061-5073. doi:10.1021/jacs.2c10547.
- Martini, M. A., Bikbaev, K., Pang, Y., Lorent, C., Wiemann, C., Breuer, N., Zebger, I., DeBeer, S., Span, I., Bjornsson, R., Birrell, J. A., Rodriguez-Macia, P. (2023). Binding of exogenous cyanide reveals new active-site states in [FeFe] hydrogenases. Chemical Science, (14) 2826-2838. doi:10.1039/d2sc06098a.
- Yogendra, S., Wilson, D. W. N., Hahn, A. W., Weyhermüller, T., Van Stappen, C., Holland, P., DeBeer, S. (2023). Sulfur-Ligated [2Fe-2C] Clusters as Synthetic Model Systems for Nitrogenase. INORGANIC CHEMISTRY, 62(6), 2663-2671. doi:10.1021/acs.inorgchem.2c03693.
- Anandaraj, S. J. L., Kang, L., DeBeer, S., Bordet, A., Leitner, W. (2023). Catalytic Hydrogenation of CO2 to Formate Using Ruthenium Nanoparticles Immobilized on Supported Ionic Liquid Phases. Small,(19) 2206806, pp. 1-10. doi:10.1002/smll.202206806.
- Caserta, G., Hartmann, S., Van Stappen, C., Karafoulidi-Retsou, C., Lorent, C., Yelin, S., Keck, M., Schoknecht, J., Sergueev, I., Yoda, Y., Hildebrandt, P., Limberg, C., DeBeer, S., Zebger, I., Frielingsdorf, S., Lenz, O. (2023). Stepwise assembly of the active site of [NiFe]-hydrogenase. Nature Chemical Biology,(19) 498-506. doi:10.1038/s41589-022-01226-w.
- Cutsail III, G., Schott-Verdugo, S., Müller, L., DeBeer, S., Groth, G., Gohlke, H. (2022). Spectroscopic and QM/MM studies of the Cu(I) binding site of the plant ethylene receptor ETR1. BIOPHYSICAL JOURNAL, 121(20), 3862-3873. doi:10.1016/j.bpj.2022.09.007.
- Keilwerth, M., Mao, W., Venturinelli Jannuzzi, S. A., Grunwald, L., Heinemann, F. W., Scheurer, A., Sutter, J., De Beer, S., Munz, D.,Meyer, K., (2023). From Divalent to Pentavalent Iron Imido Complexes and an Fe(V) Nitride via N-C Bond Cleavage. Journal of the American Chemical Society, (145), 873-887. doi:10.1021/jacs.2c09072.
- Bowker, M., DeBeer, S., Dummer, N. F., Hutchings, G. J., Scheffler, M., Schüth, F., Taylor, S., Tueysuez,H. (2022). Advancing Critical Chemical Processes for a Sustainable Future: Challenges for Industry and the Max Planck-Cardiff Centre on the Fundamentals of Heterogeneous Catalysis (FUNCAT). Angewandte Chemie, International Edition in English, (61): e202209016, pp. 1-13. doi:10.1002/anie.202209016.
- Alkan, B., Braun, M., Landrot, G., Rüdiger, O., Andronescu, C., DeBeer, S., Schulz, C., Wiggers, H. (2022). Spray-flame-synthesized Sr- and Fe-substituted LaCoO3 perovskite nanoparticles with enhanced OER activities. Journal of Materials Science, (57), 18923-18936. doi:10.1007/s10853-022-07738-z.
- Yu, M., Weidenthaler, C., Wang, Y., Budiyanto, E., Sahin, E. O., Chen, M., DeBeer, S., Rüdiger, O., Tueysuez, H. (2022). Surface Boron Modulation on Cobalt Oxide Nanocrystals for Electrochemical Oxygen Evolution Reaction. Angewandte Chemie, International Edition in English, (61): e202211543, pp. 1-12. doi:10.1002/anie.202211543.
- Mao, W., Fehn, D., Heinemann, F. W., Scheurer, A., van Gastel, M., Jannuzzi V, S. A., DeBeer, S., Munz, D., Meyer, K. (2022). Umpolung in a Pair of Cobalt(III) Terminal Imido/Imidyl Complexes. Angewandte Chemie International Edition, (61): e202206848, pp. 1-9. doi:10.1002/anie.202206848.
- Cutsail III, G. E., Banerjee, R., Rice, D. B., Stepanic, O. M., Lipscomb, J. D., DeBeer, S. (2022). Determination of the iron(IV) local spin states of the Q intermediate of soluble methane monooxygenase by K beta X-ray emission spectroscopy. Journal of Biological Inorganic Chemistry, (27), 573-582. doi:10.1007/s00775-022-01953-4.
- Decamps, L. B., Rice, D. B., DeBeer, S. (2022). An Fe6C Core in All Nitrogenase Cofactors. Angewandte Chemie International Edition (61) e202209190, pp. 1-3. doi:10.1002/anie.202209190.
- Cutsail III, G. E., DeBeer, S. (2022). Challenges and Opportunities for Applications of Advanced X-ray Spectroscopy in Catalysis Research. ACS Catalysis, 12(10), 5864-5886. doi:10.1021/acscatal.2c01016.
- Xiang, W., Yang, N., Li, X., Linnemann, J., Hagemann, U., Ruediger, O.,Heidelmann,M.; Falk, T., Aramani, M., DeBeer, S., Muhler, M.,Tschulik, K., Li, T.. (2022). 3D atomic-scale imaging of mixed Co-Fe spinel oxide nanoparticles during oxygen evolution reaction. Nature Communications, 13(179), 1-14. doi:10.1038/s41467-021-277.
- Aldous, L., Comba, P., DeBeer, S., Dey, A., Draksharapu, A., Duboc, C., Itoh, S., Karlin, K., Kundu, S., Lopez Domene, R., Marechal, J.-D., Mazumdar, S., Mukherjee, R., Parker, D., Pordea, A., Que, L., Rath, S. P., Sadler, P., Sastri, C., Schindler, S., Schunemann, V., Sen Gupta, S., Solomon, E. I., P. Stack, T. D. (2022). Small molecule activation and synthetic analogues: general discussion. Faraday Discussions, (234), 129-142. doi:10.1039/d2fd90012b.
- Anilkumar, A., Ash, P., Chakravarty, A. R., Comba, P., DeBeer, S., Dey, A., Draksharapu, A., Goswami, D., Itoh, S., Karlin, K., Lakshmi, K. V., Mazumdar, S., Pantazis, D., Parker, D., Que, L., Rajaraman, G., Rath, S. P., Sastri, C., Sen Gupta, S., Solomon, E. I. (2022). Electron transfer, spectroscopy and theory: general discussion. Faraday Discussions, 234(0), 245-263. doi:10.1039/d2fd90013k.
- Chatterjee, S., Harden, I., Bistoni, G., Castillo, R. G., Chabbra, S., van Gastel, M., Schnegg, A., Bill, E., Birrell, J. A., Morandi, B., Neese, F., DeBeer, S. (2022). A Combined Spectroscopic and Computational Study on the Mechanism of Iron-Catalyzed Aminofunctionalization of Olefins Using Hydroxylamine Derived N-O Reagent as the "Amino" Source and "Oxidant". Journal of the American Chemical Society, 144(6), 2637-2656. doi:10.1021/jacs.1c11083.
- Souilah, C., Venturinelli Jannuzzi, S. A., Demirbas, D., Ivlev, S., Swart, M., DeBeer, S., Casitas, A. (2022). Synthesis of Fe-III and Fe-IV Cyanide Complexes Using Hypervalent Iodine Reagents as Cyano-Transfer One-Electron Oxidants. Angewandte Chemie, International Edition in English, (61): e202201699, pp. 1-7. doi:10.1002/anie.202201699.
- Sisodiya-Amrute, S., Van Stappen, C., Rengshausen, S., Han, C., Sodreau, A., Weidenthaler, C., Tricard, S., DeBeer, S., Chaudret, B., Bordet, A., Leitner, W. (2022). <p>Bimetallic MxRu100_x nanoparticles (M = Fe, Co) on supported ionic liquid phases (MxRu100-x@SILP) as hydrogenation catalysts: Influence of M and M:Ru ratio on activity and selectivity</p>. Journal of Catalysis,(407), 141-148. doi:10.1016/j.jcat.2022.01.030.
- Czastka, K., Alsheikh Oughli, A., Rüdiger, O. ,DeBeer, S. (2022). Enzymatic X-ray Absorption Spectroelectrochemistry. Faraday Discussions, (234), 214-231. doi:10.1039/D1FD00079A.
- Henthorn, J. T., DeBeer, S. (2022). Selenium Valence-to-Core X-ray Emission Spectroscopy and K beta HERFD X-ray Absorption Spectroscopy as Complementary Probes of Chemical and Electronic Structure. Inorganic Chemistry, 61(6), 2760-2767. doi:10.1021/acs.inorgchem.1c02802.
- Van Stappen, C., Jimenez-Vicente, E., Perez-Gonzalez, A., Yang, Z.-Y., Seefeldt, L. C., DeBeer, S., Dean, D. R., Decamps, L. B. (2022). A conformational role for NifW in the maturation of molybdenum nitrogenase P-cluster. Chemical Science, 13(13), 3489-3500. doi:10.1039/d1sc06418e.
- Jedrzkiewicz, D., Mai, J., Langer, J., Mathe, Z., Patel, N., DeBeer, S., Harder, S. (2022). Access to a Labile Monomeric Magnesium Radical by Ball-Milling. Angewandte Chemie, International Edition in English, (xx): e202200511, pp. 1-7. doi:10.1002/anie.202200511.
- Levin, N., Casadevall, C., Cutsail III, G. E., Lloret-Fillol, J., DeBeer, S., Rüdiger, O. (2022). XAS and EPR in Situ Observation of Ru(V) Oxo Intermediate in a Ru Water Oxidation Complex**. CHEMELECTROCHEM, (9): e202101271, pp. 1-4. doi:10.1002/celc.202101271.
- Henthorn, J. T., Cutsail III, G. E., Weyhermüller, T., DeBeer, S. (2022). Stabilization of intermediate spin states in mixed-valent diiron dichalcogenide complexes. Nature Chemistry, (14), 328-333. doi:10.1038/s41557-021-00853-5.
- Geoghegan, B. L., Liu, Y., Peredkov, S., Dechert, S., Meyer, F., DeBeer, S., Cutsail III, G. E. (2022). Combining Valence-to-Core X-ray Emission and Cu K-edge X-ray Absorption Spectroscopies to Experimentally Assess Oxidation State in Organometallic Cu(I)/(II)/(III) Complexes. Journal of the American Chemical Society, 144(6), 2520-2534. doi:10.1021/jacs.1c09505.
- Czastka, K., Alsheikh Oughli, A., Rüdiger, O., DeBeer, S. (2021). Enzymatic X-ray Absorption Spectroelectrochemistry. Faraday Discussions, (xx), 1-14. doi:10.1039/x0xx00000x.
- Gerz, I., Venturinelli Jannuzzi, S. A., Hylland, K. T., Negri, C., Wragg, D. S., Öien-Ödegaard, S.,Tilset, M., Olybye, U., DeBeer, S., Amedjkouh, M. (2021). Structural Elucidation, Aggregation, and Dynamic Behaviour of N,N,N,N-Copper(I) Schiff Base Complexes in Solid and in Solution: A Combined NMR, X-ray Spectroscopic and Crystallographic Investigation. European Journal of Inorganic Chemistry. doi:10.1002/ejic.202100982.
- Spiller, N. B., Bjornsson, R., DeBeer, S., Neese, F. (2021). Carbon Monoxide Binding to the Iron–Molybdenum Cofactor of Nitrogenase: a Detailed Quantum Mechanics/Molecular Mechanics Investigation. Inorganic Chemistry, 60(23), 18031-18047. doi:10.1021/acs.inorgchem.1c02649.
- Martini, M. A., Rüdiger, O., Breuer, N., Nöring, B., DeBeer, S., Rodriguez-Macia, P., Birrell, J. (2021). The Nonphysiological Reductant Sodium Dithionite and [FeFe] Hydrogenase: Influence on the Enzyme Mechanism. Journal of the American Chemical Society, (xx), xx-xx. doi:10.1021/jacs.1c07322.
- Budiyanto, E., Zerebecki, S., Weidenthaler, C., Kox, T., Kenmoe, S., Spohr, E., DeBeer, S., Rüdiger, O., Reichenberger, S., Barcikowski, S., Tuysuz, H., (2021). Impact of Single-Pulse, Low-Intensity Laser Post-Processing on Structure and Activity of Mesostructured Cobalt Oxide for the Oxygen Evolution Reaction. ACS applied materials & interfaces. doi:10.1021/acsami.1c08034.
- Gil-Sepulcre, M., Lindner, J. O., Schindler, D., Velasco, L., Moonshiram, D., Rüdiger, O., De Beer, S., Stepanenko, V., Solano, E., Würthner, F., Llobet, A. (2021). Surface-Promoted Evolution of Ru-bda Coordination Oligomers Boosts the Efficiency of Water Oxidation Molecular Anodes. Journal of the American Chemical Society, 143(30), 11651-11661. doi:10.1021/jacs.1c04738.
- Wang, C.-H., DeBeer, S. (2021) Structure, reactivity, and spectroscopy of nitrogenase-related synthetic and biological clusters. CHEMICAL SOCIETY REVIEWS.(50)8743-8761 doi:10.1039/d1cs00381j.
- Schulz, C. E., Castillo, R. G., Pantazis, D. A., DeBeer, S., Neese, F. (2021). Structure-Spectroscopy Correlations for Intermediate Q of Soluble Methane Monooxygenase: Insights from QM/MM Calculations. JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 143(17), 6560-6577. doi:10.1021/jacs.1c01180.
- Mathe, Z., McCubbin Stepanic, O., Peredkov, S., DeBeer, S. (2021). Phosphorus K beta X-ray emission spectroscopy detects non-covalent interactions of phosphate biomolecules in situ. Chemical Science. doi:10.1039/d1sc01266e.
- Cutsail III, G. E., Ross, M. O., Rosenzweig, A. C., DeBeer, S. (2021). Towards a unified understanding of the copper sites in particulate methane monooxygenase: an X-ray absorption spectroscopic investigation dagger. Chemical Science, (12), 6194-6209. doi:10.1039/d1sc00676b.
- Gomez Castillo, R., Hahn, A. W., van Kuiken, B. E., Henthorn, J. T., McGale, J., DeBeer, S. (2021). Probing Physical Oxidation State by Resonant X-ray Emission Spectroscopy: Applications to Iron Model Complexes and Nitrogenase. Angewandte Chemie, International Edition in English, (60), 10112-10121 doi:10.1002/anie.202015669.
- Rosenbach, H., Walla, E., Cutsail III, G. E., Birrell, J. A., Pascual-Ortiz, M., DeBeer, S., Fleig, U., Span, I. (2021). The Asp1 pyrophosphatase from S. pombe hosts a [2Fe-2S]2+ cluster in vivo. Journal of Biological Inorganic Chemistry. https://doi:10.1007/s00775-020-01840-w.
- Peters, J. W., Einsle, O., Dean, D. R., DeBeer, S., Hoffman, B. M., Holland, P. L., Seefeldt, L. C. (2021). Comment on "Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase". Science, 371(6530). https://doi:10.1126/science.abe5481.
- Duan, P.-C., Schulz, R. A., Römer, A., Van Kuiken, B. E., Dechert, S., Demeshko, S., Cutsail III, G. E., DeBeer, S., Mata, R. A., Meyer, F. (2021). Ligand Protonation Triggers H-2 Release from a Dinickel Dihydride Complex to Give a Doubly "T"-Shaped Dinickel(I) Metallodiradical. Angewandte Chemie, International Edition in English, 60(4), 1891-1896. https://doi:10.1002/anie.202011494.
- Rengshausen, S., Van Stappen, C., Levin, N., Tricard, S., Luska, K.L., DeBeer, S., Chaudret, B., Bordet, A., Leitner, W. (2021). Organometallic Synthesis of Bimetallic Cobalt‐Rhodium Nanoparticles in Supported Ionic Liquid Phases (CoxRh100−x@SILP) as Catalysts for the Selective Hydrogenation of Multifunctional Aromatic Substrates Small, 17, 2006683 (10pp) https://doi.org/10.1002/smll.202006683
- Van Stappen, C., Decamps, L., DeBeer, S. (2021). Preparation and Spectroscopic Characterization of Lyophilized Mo Nitrogenase Journal of the Biological Inorganic Chemistry. 26:81–91 https://doi.org/10.1007/s00775-020-01838-4
- Rodríguez-Maciá, P., Breuer, N., DeBeer, S., Birrell, J.A. (2020). Insight into the Redox Behavior of the [4Fe–4S] Subcluster in [FeFe] Hydrogenases ACS Catalysis 10(21), 13084-13095. https://doi.org/10.1021/acscatal.0c02771
- McCubbin Stepanic, O., Ward, J., Penner-Hahn, J.E., Deb, A., Bergmann, U., DeBeer, S. (2020). Probing a silent metal: A Combined X-ray Absorption and Emission Spectroscopic Study of Biologically Relevant Zinc Complexes Inorganic Chemistry. 59, 18, 13551–13560 https://doi.org/10.1021/acs.inorgchem.0c01931
- Jensen, K.M. Ø., DeBeer, S., Koziej, D. (2020). Editorial: Spectroscopy and scattering for chemistry: new possibilities and challenges with large scale facilities Nanoscale 12(35), 17968-17970. https://doi.org/10.1039/D0NR90182B
- Zimmermann, P., Peredkov, S., Abdala P.M., DeBeer, S., Tromp, M., Müller, C., van Bokhoven, J.A. (2020). Modern X-ray spectroscopy: XAS and XES in the laboratory Coordination Chemistry Reviews 423, 213466. https://doi.org/10.1016/j.ccr.2020.213466
- Budiyanto, E., Yu, M., Chen, M., DeBeer, S., Rüdiger, O., Tüysüz, H. (2020). Tailoring Morphology and Electronic Structure of Cobalt Iron Oxide Nanowires for Electrochemical Oxygen Evolution Reaction ACS Applied Energy Materials 3(9), 8583-8594. https://doi.org/10.1021/acsaem.0c01201
- Beheshti Askari, A., al Samarai, M., Hiraoka, N., Ishii, H., Tillmann, L., Muhler, M., DeBeer, S. (2020). In situ X-ray emission and high-resolution X-ray absorption spectroscopy applied to Ni-based Bimetallic Dry Methane Reforming Catalysts Nanoscale 20(28), 15185-15192. https://doi.org/10.1039/D0NR01960G
- Yu, M., Moon, G.-H., Castillo, R.G., DeBeer, S., Weidenthaler, C., Tüysüz, H. (2020). Dual Role of Silver Moieties Coupled with Ordered Mesoporous Cobalt Oxide towards Electrocatalytic Oxygen Evolution Reaction Angewandte Chemie International Edition 59(38), 16544-16552. https://doi.org/10.1002/anie.202003801
- DeBeer, S. (2020). Introduction to X-ray spectroscopy – including X-ray absorption, X-ray emission and resonant inelastic X-ray scattering Bioorganometallic Chemistry 407-432. https://doi.org/10.1515/9783110496574-011
- Rodríguez-Maciá, P., Galle, L., Bjornsson, R., Lorent, C., Zebger, I., Yoda, Y., Cramer, S., DeBeer, S., Span, I., Birrell, J.A. (2020). Caught in the Hinact: Crystal Structure and Spectroscopy Reveal a Sulfur Bound to the Active Site of an O2‐stable State of [FeFe] Hydrogenase Angewandte Chemie International Edition 59(38), 16786-16794. https://doi.org/10.1002/anie.202005208
- Levin, N., Peredkov, S., Weyhermüller, T., Rüdiger, O., Pereira, N.B., Grötzsch, D., Kalinko, A., DeBeer, S. (2020). Ruthenium 4d-to-2p X-ray Emission Spectroscopy: A Simultaneous Probe of the Metal and the Bound Ligands Inorganic Chemistry 59(12), 8272-8283. https://doi.org/10.1021/acs.inorgchem.0c00663
- Castillo, R.G., Henthorn, J.T., McGale, J., Maganas, D., DeBeer, S. (2020). Kβ X‐ray Emission Spectroscopic study of a second‐row transition metal (Mo) and its application to nitrogenase related model complexes Angewandte Chemie International Edition 59(31), 12965-12975. https://doi.org/10.1002/anie.202003621
- Beheshti-Askari. A., al Samarai, M., Morana, B., Tillmann, L., Pfänder, N., Wandzilak, A., Watts, B., Belkhou, R., Muhler, M., DeBeer, S. (2020). In-situ X-ray Microscopy reveals particle dynamics in a NiCo dry methane reforming catalyst under operating conditions ACS Catalysis 10(11), 6223-6230. https://doi.org/10.1021/acscatal.9b05517
- Van Stappen, C., Decamps, L., Cutsail III, G.E., Bjornsson, R., Henthorn, J.T., Birrell, J.A., DeBeer, S. (2020). The Spectroscopy of Nitrogenases Chemical Reviews 120(12), 5005-5081. https://doi.org/10.1021/acs.chemrev.9b00650
- Spiller, N., Chilkuri, V.G., DeBeer, S., Neese, F. (2020). Sulfur vs. Selenium as Bridging Ligand in Di‐Iron Complexes: A Theoretical Analysis European Journal of Inorganic Chemistry 2020(15-16), 1525-1538. https://doi.org/10.1002/ejic.202000033
- Maganas, D., Kowalska, J.K., Van Stappen, C., DeBeer, S., Neese, F. (2020). Mechanism of L2,3-edge X-Ray Magnetic Circular Dichroism Intensity from Quantum Chemical Calculations and Experiment - A case study on V(IV)/V(III) complexes The Journal of Chemical Physics 152(11), 114107. https://doi.org/10.1063/1.5129029
- Cutsail III, G.E., Blaesi, E.J., Pollock, C.J., Bollinger Jr, J.M., Krebs, C., DeBeer, S. (2020). High-resolution iron X-ray absorption spectroscopic and computational studies of non-heme diiron peroxo intermediates Journal of Inorganic Biochemistry 203, 110877. https://doi.org/10.1016/j.jinorgbio.2019.110877
- Birrell, J.A., Pelmenschikov, V., Mishra, N., Wang, H., Yoda, Y., Tamasaku, K., Rauchfuss, T.B., Cramer, S.P., Lubitz, W., DeBeer, S. (2020). Spectroscopic and Computational Evidence that [FeFe] Hydrogenases Operate Exclusively with CO-bridged Intermediates Journal of the American Chemical Society 142(1), 222-232. https://doi.org/10.1021/jacs.9b09745
- Chilkuri, V.G., DeBeer, S., Neese, F. (2020). Ligand Field Theory and Angular Overlap Model Based Analysis of the Electronic Structure of Homovalent Iron–Sulfur Dimers Inorganic Chemistry 59(2), 984-995. https://doi.org/10.1021/acs.inorgchem.9b00974
- Liu, Y., Resch, S.G., Klawitter, I., Cutsail III, G.E., Demeshko, S., Dechert, S., Kühn, F.E., DeBeer, S., Meyer, F. (2020). An Adaptable N‐Heterocyclic Carbene Macrocycle Hosting Copper in three Oxidation States Angewandte Chemie International Edition 59(14), 5696-5705. https://doi.org/10.1002/anie.201912745
- Mathe, Z., Pantazis, D.A., Lee, H.B., Gnewkow, R., Van Kuiken, B., Agapie, T., DeBeer, S. (2019). Calcium Valence-to-Core X-ray Emission Spectroscopy: A Sensitive Probe of Oxo Protonation in Structural Models of the Oxygen-Evolving Complex Inorganic Chemistry 58(23), 16292-16301. https://doi.org/10.1021/acs.inorgchem.9b02866
- DeRosha, D.E., Chilkuri, V.G., Van Stappen, C., Bill, E., Mercado, B.Q., DeBeer, S., Neese, F., Holland, P.L. (2019). Planar three-coordinate iron sulfide in a synthetic [4Fe-3S] cluster with biomimetic reactivity Nature Chemistry 11, 1019–1025. https://doi.org/10.1038/s41557-019-0341-7
- McGale, J., Cutsail, G.E. III, Joseph, C., Rose, M.J., DeBeer, S. (2019). Spectroscopic X-ray and Mössbauer Characterization of M6 and M5 Iron(Molybdenum)-Carbonyl Carbide Clusters: High Carbide-Iron Covalency Enhances Local Iron Site Electron Density Despite Cluster Oxidation Inorganic Chemistry 58(19), 12918-12932. https://doi.org/10.1021/acs.inorgchem.9b01870
- Al Samarai, M., Hahn, A.W., Askari, A.B., Cui, Y.-T., Yamazoe, K., Miyawaki, J., Harada, Y., Rüdiger, O., DeBeer, S. (2019). Elucidation of Structure-Activity Correlations in a Nickel-Manganese Oxide OER Catalyst by Operando Ni L-edge XAS and 2p3d RIXS ACS Applied Materials and Interfaces 11(42), 38595-38605. https://doi.org/10.1021/acsami.9b06752
- Van Stappen, C., Thorhallsson, A.T., Decamps, L., Bjornsson, R., DeBeer, S. (2019). Resolving the structure of the E1 state of Mo Nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations Chemical Science 10(42), 9807-9821. https://doi.org/10.1039/c9sc02187f
- Van Stappen, C., Davydov, R., Yang, Z.-Y., Fan, R., Guo, Y., Bill, E., Seefeldt, L.C., Hoffman, B.M., DeBeer, S. (2019). A spectroscopic description of the E1 state of Mo Nitrogenase based on Mo and Fe X-ray absorption and Mössbauer studies Inorganic Chemistry 58(18), 12365-12376. https://doi.org/10.1021/acs.inorgchem.9b01951
- Speelman, A.L., Čorić, I., Van Stappen, C., DeBeer, S., Mercado, B.Q., Holland, P.L. (2019). Nitrogenase-Relevant Reactivity of a Synthetic Iron–Sulfur–Carbon Site Journal of the American Chemical Society 141(33), 13148-13157. https://doi.org/10.1021/jacs.9b05353
- Chrysina, M., Heyno, E., Kutin Y., Reus, M., Nilsson, H., Nowaczyk. M.N., DeBeer, S., Neese, F., Messinger, J., Lubitz, W., Cox, N. (2019). Five-coordinate MnIV intermediate in the activation of nature’s water splitting cofactor Proceedings of the National Academy of Sciences of the United States of America 116(34), 16841-16846. https://doi.org/10.1073/pnas.1817526116
- Henthorn, J.T., Arias, R.J., Koroidov, S., Kroll, T., Sokaras, D., Bergmann, U., Rees, D.C., DeBeer, S. (2019). Localized Electronic Structure of Nitrogenase FeMoco Revealed by Selenium K-edge High Resolution X-ray Absorption Spectroscopy Journal of the American Chemical Society 141(34), 13676-13688. https://doi.org/10.1021/jacs.9b06988
- Yogendra, S., Weyhermüller, T., Hahn, A.W., DeBeer, S. (2019). From Ylides to Doubly Yldiide-Bridged Iron(II) High Spin Dimers via Self-Protolysis Inorganic Chemistry 58(14), 9358-9367. https://doi.org/10.1021/acs.inorgchem.9b01086
- Kowalska, J.K., Henthorn, J.T., Van Stappen, C., Trncik, C., Einsle, O., Keavney, D., DeBeer, S. (2019). X-ray Magnetic Circular Dichroism Spectroscopy Applied to Nitrogenase and Related Models: Experimental Evidence for a Spin-Coupled Mo(III) Angewandte Chemie International Edition 58(28), 9373-9377. https://doi.org/10.1002/anie.201901899
- Cutsail III, G.E., Gagnon, N.L., Spaeth, A.D., Tolman, W.B., DeBeer, S. (2019). Valence‐to‐Core X‐ray Emission Spectroscopy as a Probe of O‐O Bond Activation in Cu2O2 complexes Angewandte Chemie International Edition 58(27), 9114-9119. https://doi.org/10.1002/anie.201903749
- Kalläne, S.I., Hahn, A.W., Weyhermüller, T., Bill, E., Neese, F., DeBeer, S., van Gastel, M. (2019). Spectroscopic and Quantum Chemical Investigation of Benzene-1,2- dithiolate-Coordinated Diiron Complexes with Relevance to Dinitrogen Activation Inorganic Chemistry 58(8), 5111-5125. https://doi.org/10.1021/acs.inorgchem.9b00177
- Maganas, D., Kowalska, J.K., Noiijen, M., DeBeer, S., Neese, F. (2019). Comparison of Multireference Ab initio Wavefunction Methodologies for X-Ray Absorption Edges: A Case study on [Fe(II/III)Cl4]2-/1- molecules The Journal of Chemical Physics 150(10), 104106. https://doi.org/10.1063/1.5051613
- Malzer, W., Grötzsch, D., Gnewkow, R., Schlesiger, C., Kowalewski, F., Van Kuiken, B., DeBeer, S., Kanngießer, B. (2018). A laboratory spectrometer for high throughput X-ray emission spectroscopy in catalysis research Review of Scientific Instruments 89, 113111. https://doi.org/10.1063/1.5035171
- Cutsail III, G.E., Banerjee, R., Zhou, A., Que, L., Lipscomb, J.D., DeBeer, S. (2018). High-Resolution EXAFS Provides Evidence for a Longer Fe•••Fe Distance in the Q Intermediate of Methane Monooxygenase Journal of American Chemical Society 140(48) 16807-16820. https://doi.org/10.1021/jacs.8b10313
- Hahn, A.W., Van Kuiken, B.E., Chilkuri, V.G., Levin, N., Bill, E., Weyhermüller, T., Nicolaou, A., Miyawaki, J., Harada, Y., DeBeer, S. (2018). Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X ray Scattering (RIXS) Inorganic Chemistry 57(37), 11918-11923. https://doi.org/10.1021/acs.inorgchem.8b01550
- Rodriguez-Maciá, P., Reijerse, E.J., van Gastel, M., DeBeer, S., Lubitz, W., Rüdiger, O., Birrell, J.A. (2018). Sulfide Protects [FeFe] Hydrogenases From O2 Journal of the American Chemical Society 140(30), 9346-9350. https://doi.org/10.1021/jacs.8b04339
- Chantzis, A., Kowalska, J.K., Maganas, D., DeBeer, S., Neese, F. (2018). Ab initio Wavefunction-based Determination of Element Specific Shifts for the Efficient Calculation of X-Ray Absorption Spectra of Main Group Elements and First Row Transition Metals Journal of Chemical Theory and Computation 14(7), 3686-3702. https://doi.org/10.1021/acs.jctc.8b00249
- Galle, L.M., Cutsail, G.E. III, Nischwitz, V., DeBeer, S., Span, I. (2018). Spectroscopic characterization of the Co-substituted C-terminal domain of rubredoxin-2 Biological Chemistry 399(7), 787-798. https://doi.org/10.1515/hsz-2018-0142
- Van Kuiken, B.E., Hahn, A.W., Nayyar, B., Schiewer, C.E., Lee, S.C., Meyer, F., Weyhermüller, T., Nicolaou, A., Cui, Y-T., Miyawaki, J., Hatada, Y., DeBeer, S. (2018). Electronic Spectra of Iron-Sulfur Complexes Measured by 2p3d RIXS Spectroscopy Inorganic Chemistry 57(12), 7355-7361. https://doi.org/10.1021/acs.inorgchem.8b01010
- Van Stappen, C., Maganas, D., DeBeer, S., Bill, E., Neese, F. (2018). Investigation of the Magnetic and Spectroscopic Properties of V(III) and V(IV) Complexes Inorganic Chemistry 57(11), 6421-6438. https://doi.org/10.1021/acs.inorgchem.8b00486
- Maganas, D., DeBeer, S., Neese, F. (2018).A Pair Natural Orbitals Restricted Open Shell Configuration Interaction (PNO-ROCIS) Approach for Calculating X-ray Absorption Spectra of Large Chemical Systems Journal of Physical Chemistry A 122(5), 1215-1227. https://doi.org/10.1021/acs.jpca.7b10880
- Leipzig, B.K., Rees, J.A., Kowalska, J.K., Theisen, R.M., Kavčič, M., Poon, P.C.Y., Kaminsky, W., DeBeer, S., Bill, E., Kovacs, J.A. (2018). How Do Ring Size and π-Donating Thiolate Ligands Affect Redox-Active, α-Imino-N-heterocycle Ligand Activation? Inorganic Chemistry 57(4), 1935-1949. https://doi.org/10.1021/acs.inorgchem.7b02748
- DeBeer, S. (2018). Advanced X-ray Spectroscopic Methods for Studying Iron-Sulfur-Containing Proteins and Model Complexes Methods Enzymology. Fe-S Cluster Enzymes Part B 599, 427-450. https://doi.org/10.1016/bs.mie.2017.09.008
- Römelt, C., Song, J.S., Tarrago, M., Rees, J.A., van Gastel, M., Weyhermüller, T., DeBeer, S., Bill, E., Neese, F., Ye, S. (2017). Electronic Structure of a Formal Iron(0), Porphyrin Complex Relevant to CO2 Reduction Inorganic Chemistry 56(8), 4745-4750. https://doi.org/10.1021/acs.inorgchem.7b00401
- Rees, J.A., Bjornsson, R., Kowalska, J.K., Lima, F.A., Schlesier, J., Sippel, D., Weyhermüller, T., Einsle, O., Kovacs, J.A., DeBeer, S. (2017). Comparative electronic structures of nitrogenase FeMoco and FeVco Dalton Transactions 46(8), 2445-2455. https://doi.org/10.1039/c7dt00128b
- Bjornsson, R., Neese, F., DeBeer, S. (2017). Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge Inorganic Chemistry 56(3), 1470-1477. https://doi.org/10.1021/acs.inorgchem.6b02540
- Castillo, R.G., Banerjee, R., Allpress, C.J., Rohde, G.T., Bill, E., Que Jr., L., Lipscomb, J.D., DeBeer, S. (2017). High-Energy-Resolution Fluorescence-Detected X-ray Absorption of the Q Intermediate of Soluble Methane Monooxygenase Journal of the American Chemical Society 139(49), 18024-18033. https://doi.org/10.1021/jacs.7b09560
- Maganas, D., DeBeer, S., Neese, F. (2017). A Restricted Open Configuration Interaction with Singles Method To Calculate Valence-to-Core Resonant X-ray Emission Spectra: A Case Study Inorganic Chemistry 56(19), 11819-11836. https://doi.org/10.1021/acs.inorgchem.7b01810
- Koziej, D., DeBeer, S. (2017). Application of Modern X-ray Spectroscopy in Chemistry-Beyond Studying the Oxidation State Chemistry of Materials 29(17), 7051-7053. https://doi.org/10.1021/acs.chemmater.7b03455
- Chilkuri, V.G., DeBeer, S., Neese, F. (2017). Revisiting the Electronic Structure of FeS Monomers Using ab Initio Ligand Field Theory and the Angular Overlap Model Inorganic Chemistry 56(17), 10418-10436. https://doi.org/10.1021/acs.inorgchem.7b01371
- Kowalska, J.K., Nayyar, B., Rees, J.A., Schiewer, C.E., Lee, S.C., Kovacs, J.A., Meyer, F., Weyhermüller, T., Otero, E., DeBeer, S. (2017). Iron L2,3-edge X-ray Absorption and X-ray Magnetic Circular Dichroism Studies of Molecular Iron Complexes with Relevance to the FeMoco and FeVco Active Sites of Nitrogenase Inorganic Chemistry 56(14), 8147-8158. https://doi.org/10.1021/acs.inorgchem.7b00852
- Hahn, A.W., Van Kuiken, B.E., al Samarai, M., Atanasov, M., Weyhermüller, T., Cui, Y.T., Miyawaki, J., Harada, Y., Nicolaou, A., DeBeer, S. (2017). Measurement of the Ligand Field Spectra of Ferrous and Ferric Iron Chlorides Using 2p3d RIXS Inorganic Chemistry 56(14), 8203-8211. https://doi.org/10.1021/acs.inorgchem.7b00940
- Casitas, A., Rees, J.A., Goddard, R., Bill, E., DeBeer, S., Fürstner, A. (2017). Two Exceptional Homoleptic Iron(IV) Tetraalkyl Complexes Angewandte Chemie International Edition 56(34), 10108-10113. https://doi.org/10.1002/anie.201612299
- Van Kuiken, B.E., Hahn, A.W., Maganas, D., DeBeer, S. (2016). Measuring Spin-Allowed and Spin-Forbidden d-d Excitations in Vanadium Complexes with 2p3d Resonant Inelastic X-ray Scattering Inorganic Chemistry 55(21), 11497-11501. https://doi.org/10.1021/acs.inorgchem.6b02053
- Kowalska, J.K., Lima, F.A., Pollock, C. J., Rees, J.A., DeBeer, S. (2016). A Practical Guide to High-resolution X-ray Spectroscopic Measurements and their Applications in Bioinorganic Chemistry Israel Journal of Chemistry 56(9-10), 803-815. https://doi.org/10.1002/ijch.201600037
- Rees, J.A., Wandzilak, A., Maganas, D., Wurster, N.I.C., Hugenbruch, S., Kowalska, J.K., Pollock, C.J., Lima, F.A., Finkelstein, K.D., DeBeer, S. (2016). Experimental and theoretical correlations between vanadium K-edge X-ray absorption and Kß emission spectra Journal of Biological Inorganic Chemistry 21(5-6), 793-805. https://doi.org/10.1007/s00775-016-1358-7
- Kupper, C., Rees, J.A., Dechert, S., DeBeer, S., Meyer, F. (2016). Complete Series of {FeNO}8, {FeNO}7, and {FeNO}6 Complexes Stabilized by a Tetracarbene Macrocycle Journal of the American Chemical Society 138(25), 7888-7898. https://doi.org/10.1021/jacs.6b00584
- Kowalska, J.K., Hahn, A.W., Albers, A., Schiewer, C.E., Bjornsson, R., Lima, F.A., Meyer, F., DeBeer, S. (2016). X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2]n Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron-Sulfur Clusters Inorganic Chemistry 55(9), 4485-4497. https://doi.org/10.1021/acs.inorgchem.6b00295
- Hugenbruch, S., Shafaat, H.S., Krämer, T., Delgado-Jaime, M.U., Weber, K., Neese, F., Lubitz, W., DeBeer, S. (2016). In search of metal hydrides: an X-ray absorption and emission study of [NiFe] hydrogenase model complexes Physical Chemistry Chemical Physics 18(16), 10688-10699. https://doi.org/10.1039/c5cp07293j
- Martin-Diaconescu, V., Chacon, K.N., Delgado-Jaime, M.U., Sokaras, D., Weng, T.C., DeBeer, S., Blackburn, N.J. (2016). Kß Valence to Core X-ray Emission Studies of Cu(I) Binding Proteins with Mixed Methionine - Histidine Coordination. Relevance to the Reactivity of the M- and H-sites of Peptidylglycine Monooxygenase Inorganic Chemistry 55(7), 3431-3439. https://doi.org/10.1021/acs.inorgchem.5b02842
- DeBeer, S., Bergmann U. (2016). X-ray Emission Spectroscopic Techniques in Bioinorganic Applications Encyclopedia of Inorganic and Bioinorganic Chemistry 1-14. https://doi.org/10.1002/9781119951438.eibc2158
Awards
- 2023 Glenn T. Seaborg Lectureship, University of California Berkeley
- 2022 R.J.P. Williams Lectureship, Oxford University
- 2022 Malcom H. Chisholm Lecturer, The Ohio State University
- 2021 Fellow of the Royal Society of Chemistry
- 2019 European Research Council Synergy Grant Awardee
- 2016 Inorganic Chemistry Lectureship Award
- 2015 Society of Biological Inorganic Chemistry, Early Career Award
- 2013 European Research Council Consolidator Grant Awardee
- 2012 Kavli Fellow, U.S. National Academy of Sciences
- 2011-2013 Alfred P. Sloan Research Fellow
Group members
Office
Valerie EstersGroup leaders
Dr. Sergey PeredkovDr. Christina Römelt
Dr. Olaf Rüdiger
Dr. Kushal Sengupta
Dr. Thomas Weyhermüller
Project leaders
Dr. Yves KayserDr. Sergio Augusto Venturinelli Jannuzzi







Inorganic Spectroscopy

Our department focuses on the development and application of advanced X-ray spectroscopic tools for understanding processes in biological and chemical catalysis. We are interested in the development of both static and time-dependent probes of transition metal electronic structure, and in particular the development of two-dimensional X-ray spectroscopic probes, which enable enhanced selectivity. Challenging questions in energy research motivate our research. Namely how can earth-abundant base metals enable the activation of strong chemical bonds? In this context, we focus on four transition-metal catalyzed reactions:
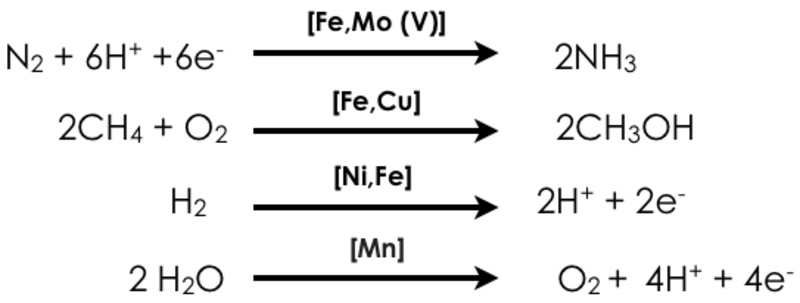

In all of the reactions shown above, there are both biological (homogenous) and heterogeneous catalysts, which are able to effect these conversions, albeit with varied efficiencies and in some cases only in multiple steps (e.g. industrial methane oxidation). We are interested in understanding the electronic structure of the catalysts and the transformations, which occur over the course of a reaction. Ideally this knowledge will form a fundamental basis for rational catalytic design. Our approach utilizes X-ray based spectroscopies, including resonant (RXES, RIXS) and non-resonant X-ray emission (XES), X-ray absorption (XAS), X-ray magnetic circular dichroism (XMCD), and nuclear resonant vibrational spectroscopy (NRVS) (Figure 1). In addition to our synchrotron-based studies, we are developing instrumentation for in-house X-ray spectroscopy. The applied combination of spectroscopic approaches provides insight into metal oxidation states, spin states, and the local coordination environments of the transition metal active sites. Our spectroscopic studies are coupled to more standard laboratory spectroscopy (UV-Vis, EPR, Raman and Mössbauer) and modern computational methods, in order to obtain quantitative insights into electronic structural changes in catalytic systems.



1. Spectroscopy Developments
1.1. Valence XES. Recent work in our research group has focused on developing the full information content of valence XES and applying this method to biological catalysts.1-12 In a valence XES measurement, one first ionizes a metal 1s electron and then monitors the resultant fluorescence after a valence electron refills the metal 1s core hole. As such, valence XES spectra provide a map of the ligand ionization potentials, and show a strong dependence on both ligand identity (i.e. C, N, or O) and protonation state (O2-, OH-, or H2O) (Figure 2). A notable application of this method was the use of valence XES to identify central carbon atom in the FeMoco cluster of nitrogenase.8 As the identification of the central atom had eluded characterization by other experimental approaches for almost a decade, this result serves to highlight the power and selectivity of the method. More recent studies in my group have focused on the use of valence XES to understand metallocofactor cluster biosynthesis and the atomic composition of FeVco in Vanadium nitrogenase.7, 13, 14
Valence XES is sensitive not only to the identity of ligand, but also to the degree of activation (i.e. weakening) of a bound ligand. For instance, when performing valence XES on a complex with an Fe-N2 moiety, spectral features which can be correlated with the N2 2s2s σ* orbital interacting with the Fe centers have been identified.12 As the N-N bond is lengthened, the energy of this transition decreases in energy, with a shift of more than 2 eV on going from an N-N triple bond to a cleaved nitride. This means that Fe valence XES is an ideal spectral probe of N-N bond cleavage (or cleavage of other diatomic ligands) by Fe (or any other transition metal).
To this end, we are developing dispersive valence XES, as a time-resolved probe of transition metal catalysis. Recently, funded by an ERC Consolidator Grant (N2RED, PI: DeBeer) and in collaboration with the group of Prof. Kangießer at TU Berlin, an in-house dispersive XES setup has been designed and commissioned. The instrument utilizes a laboratory X-ray source (Excillum, Metal Jet) in combination with a cylindrical Highly Annealed Pyrolytic Graphite (HAPG) crystal and a CCD detector to obtain XES in the 2.4-9 keV range (Figure 3). This unique in-house setup helps ease demands on synchrotron time, providing an ideal setup for concentrated samples and a means to obtain preliminary measurements before conducting more complex synchrotron or X-ray free electron laser measurements. In the context of synchrotron-based dispersive XES, the Department of Inorganic Spectroscopy is also leading development of the PINK beam line at the Energy Materials In-situ Laboratory in Berlin. PINK will provide an intense source for simultaneous “two-color” XES measurements of heterometallic catalysts. These efforts are lead by group leader Dr. Sergey Peredkov.
1.2 Resonant valence XES. To further enhance the selectivity of valence XES, we are developing resonant valence XES.2 This is a two-dimensional measurement where one resonantly excites a metal 1s core electron into the partially occupied and unoccupied states localized on the photoabsorber (i.e. the equivalent of an X-ray absorption edge scan), while simultaneously monitoring the highest energy emission processes (i.e. the valence XES). In doing so, one is able to construct a two-dimensional plane, with the incident energy on the x-axis and the emission on the y-axis (Figure 4). The horizontal cuts through this plane, then represent the XAS data at a given emission energy and effectively correspond to “ligand selective XAS”. While the vertical cuts represent the XES data at a given incident energy or valence RXES. We believe this approach should be of particular utility for detecting the XAS of different types of metal-ligand interactions within complex media.
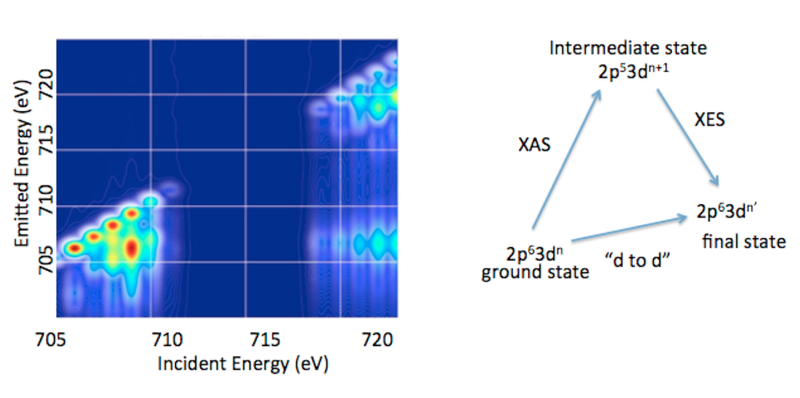
1.3. 2p3d RIXS. In addition to valence RXES, we are developing the full information content of transition metal 2p3d RXES or what is more commonly referred to as 2p3d RIXS.15 2p3d RIXS involves the dipole allowed excitation of a 2p63dn ground state to arrive at a2p53dn+1 intermediate state, which subsequently decays via a dipole allowed process to a 2p63dn’final state (Figure 5). Hence the difference between the ground state and the final state corresponds to formal dipole forbidden d-to-d transitions, which one arrives at through two dipole allowed processes. In a 2p3d RIXS measurement, the energy difference between the ground state and the final state is referred to as the “energy transfer” axis, which due to the nature of the experiment may be readily measured from 0-10 eV (or from 0 to over 80,000 cm-1). This means that spectral regions that cannot be accessed using standard optical or infrared spectroscopic probes are accessible. In addition, 2p3d RIXS experiments have been shown, in certain cases, to allow for the observation of formally spin forbidden transitions due to intermediate state spin orbit coupling. Hence, it may be possible to effectively measure the complete spin ladder, thus providing unprecedented experimental insights into transition metal electronic structure.
2. Applications
The spectroscopy developments discussed in the proceeding sections form a strong basis for applications in biological and chemical catalysis. In the section that follows, we briefly highlight some of our ongoing and planned efforts.
N2 reduction. The transformation of N2 to ammonia (NH3) is crucial for life on earth. This kinetically challenging processes is enabled biologically by the enzyme nitrogenase and industrially by the Haber-Bosch process, with remarkable efficiencies. In contrast, chemists have yet to synthesize a molecular system that can compete with either the biological or the industrial process. It is our view that inability to design effective molecular catalysts derives in part from a lack of fundamental knowledge. How does one effectively cleave N2, the strongest homodiatomic bond in known in chemistry? How does the metal catalyst interact with N2? What intermediate species are generated in the process of N2 bond breaking? To answer these questions we are applying advanced x-ray spectroscopic methods to both the biological and the heterogeneous catalysts. For the biological catalyst, we seek to understand the complex electronic structure of the ground state, the role of the central carbon, the role of the heterometal (Mo or V) and the site of substrate interaction. For the heterogeneous catalysts, we seek to understand the nature of the surface nitrides, the oxidation state of the “active” Fe and the interaction of the potassium promoter. These questions will be answered using the advanced spectroscopic approaches detailed in the preceding sections.
CH4 Oxidation. The biological oxidation of methane to methanol by soluble methane monoxygenase is a process of fundamental interest in the context of a sustainable energy economy. Presently we are focusing on elucidating the mechanism of biological methane oxidation, and in particular determining the controversial structure of the intermediate MMO-Q, which is the species responsible for extracting a proton from inert methane. Utilizing high-resolution XAS methods, we have recently obtained new insights into the elextronic structure of MMO-Q. This motivates further freeze-quench, as well as in situ studies, of biological methane oxidation.
H2O Oxidation. Similar to our studies of N2 reduction, we are also interested in understanding the process of Mn-mediated water oxidation at both the biological and heterogeneous limits. To this end, we utilize advanced X-ray spectroscopic methods to examine Mn birnessites, surface doped Mn systems, and the Mn4Ca active site of photosystem II. “Two-color” simultaneous Mn and Ca XES will be utilized in a time-resolved fashion to evaluate the role of both the Mn and the Ca in catalysis. Our goal is to obtain atomic level insights into the factors that govern O-O bond formation.
H2 production. Hydrogen production and activation has been the focus of extensive research over the last few decades in the context of renewable energy and carbon-neutral fuels. The most prevalent and robust hydrogenases contain a heterobimetallic [NiFe] active site. The redox-active nickel center is thought to play a critical role in hydrogen binding and activation, and many prior studies using x-ray crystallography, electrochemistry, FTIR, and EPR spectroscopy have been carried out to characterize the activation and mechanism of the enzyme. However, of the three catalytically relevant states, only the Ni-C state is paramagnetic. Thus, along with the aerobically-inactivated, hydroxide-bridged Ni-B state, the hydride-bridged Ni-C state represents the only catalytic intermediate that has been structurally well-characterized. Many approaches have been attempted to characterize the other two intermediates, the Ni-SI and Ni-R states, including FTIR and X-ray absorption spectroscopy. However, a number of questions remain, including identification of the nickel spin states, nature of the bridging ligand between the two metal centers in each redox state, metal to which dihydrogen coordinates, and mode of hydrogen binding to that metal. These questions will be addressed through a combination of advanced X-ray based approaches and should provide fundamental insights into the mechanism of biological H2 production.
References
[1] Delgado-Jaime, M. U., Dible, B. R., Chiang, K. P., Brennessel, W. W., Bergmann, U., Holland, P. L., and DeBeer, S. (2011) Identification of a Single Light Atom within a Multinuclear Metal Cluster Using Valence-to-Core X-ray Emission Spectroscopy, Inorg Chem 50, 10709-10717.
[2] Hall, E. R., Pollock, C. J., Bendix, J., Collins, T. J., Glatzel, P., and DeBeer, S. (2014) Valence-to-Core-Detected X-ray Absorption Spectroscopy: Targeting Ligand Selectivity, J Am Chem Soc 136, 10076-10084.
[3] Hugenbruch, S., Shafaat, H. S., Kramer, T., Delgado-Jaime, M. U., Weber, K., Neese, F., Lubitz, W., and DeBeer, S. (2016) In search of metal hydrides: an X-ray absorption and emission study of [NiFe] hydrogenase model complexes, Phys Chem Chem Phys 18, 10688-10699.
[4] Kowalska, J., and DeBeer, S. (2015) The role of X-ray spectroscopy in understanding the geometric and electronic structure of nitrogenase, Bba-Mol Cell Res 1853, 1406-1415.
[5] Kowalska, J. K., Hahn, A. W., Albers, A., Schiewer, C. E., Bjornsson, R., Lima, F. A., Meyer, F., and DeBeer, S. (2016) X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2](n) Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron-Sulfur Clusters, Inorg Chem 55, 4485-4497.
[6] Kowalska, J. K., Lima, F. A., Pollock, C. J., Rees, J. A., and DeBeer, S. (2016) A Practical Guide to High-resolution X-ray Spectroscopic Measurements and their Applications in Bioinorganic Chemistry, Isr J Chem 56, 803-815.
[7] Lancaster, K. M., Hu, Y. L., Bergmann, U., Ribbe, M. W., and DeBeer, S. (2013) X-ray Spectroscopic Observation of an Interstitial Carbide in NifEN-Bound FeMoco Precursor, J Am Chem Soc 135, 610-612.
[8] Lancaster, K. M., Roemelt, M., Ettenhuber, P., Hu, Y. L., Ribbe, M. W., Neese, F., Bergmann, U., and DeBeer, S. (2011) X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor, Science 334, 974-977.
[9] Lee, N., Petrenko, T., Bergmann, U., Neese, F., and DeBeer, S. (2010) Probing Valence Orbital Composition with Iron K beta X-ray Emission Spectroscopy, J Am Chem Soc 132, 9715-9727.
[10] Pollock, C. J., and DeBeer, S. (2011) Valence-to-Core X-ray Emission Spectroscopy: A Sensitive Probe of the Nature of a Bound Ligand, J Am Chem Soc 133, 5594-5601.
[11] Pollock, C. J., and DeBeer, S. (2015) Insights into the Geometric and Electronic Structure of Transition Metal Centers from Valence-to-Core X-ray Emission Spectroscopy, Accounts Chem Res 48, 2967-2975.
[12] Pollock, C. J., Grubel, K., Holland, P. L., and DeBeer, S. (2013) Experimentally Quantifying Small-Molecule Bond Activation Using Valence-to-Core X-ray Emission Spectroscopy, J Am Chem Soc 135, 11803-11808.
[13] Rees, J. A., Martin-Diaconescu, V., Kovacs, J. A., and DeBeer, S. (2015) X-ray Absorption and Emission Study of Dioxygen Activation by a Small-Molecule Manganese Complex, Inorg Chem 54, 6410-6422.
[14] Rees, J. A., Bjornsson, R., Kowalska, J. K., Lima, F. A., Schlesier, J., Sippel, D., Weyhermueller, T., Einsle, O., Kovacs, J. A., and DeBeer, S. (2017) Comparative electronic structures of nitrogenase FeMoco and FeVco, Dalton T 46, 2445-2455.
[15] Van Kuiken, B. E., Hahn, A. W., Maganas, D., and DeBeer, S. (2016) Measuring Spin-Allowed and Spin-Forbidden d-d Excitations in Vanadium Complexes with 2p3d Resonant Inelastic X-ray Scattering, Inorg Chem 55, 11497-11501.