Dr. Thomas Weyhermüller - Chemische Synthese, Röntgenstrukturanalyse, NMR

- Dr. Thomas Weyhermüller
- Gruppenleiter
- Chemische Synthese, Röntgenstrukturanalyse, NMR
- Anorganische Spektroskopie
- +49 (0)208 306 - 3652
- thomas.weyhermueller(at)cec.mpg.de
- Raum: 514
Vita
| Diplom (Chemie) | Ruhr-Universität Bochum (1989) |
| Ph.D., wiss. MA | Ruhr-Universität Bochum (1990-1994) |
| Dr. rer. nat. | Ruhr-Universität Bochum (1994) |
| Gruppenleiter | MPI für Bioanorganische Chemie; heute: MPI CEC (since 1995) |
Publications
Full publications list | ORCID | ResearcherID
Selected MPI CEC publications
- Durin, G., Lee, M., Pogany, M. A., Weyhermüller, T., Kaeffer, N., Leitner, W. (2023). Hydride-Free Hydrogenation: Unraveling the Mechanism of Electrocatalytic Alkyne Semihydrogenation by Nickel-Bipyridine Complexes. JOURNAL OF THE AMERICAN CHEMICAL SOCIETy, 145(31), 17103-17111. doi:10.1021/jacs.3c03340.
- Chang, W.-C., Randel, H., Weyhermüller, T., Auer, A. A. A., Fares, C., Werlé, C. (2023). A Cooperative Rhodium/Secondary Phosphine Oxide [Rh/P(O)nBu(2)] Template for Catalytic Hydrodefluorination of Perfluoroarenes. Angewandte Chemie, International Edition in English, (62): e202219127, pp. 1-10. doi:10.1002/anie.202219127.
- Yogendra, S., Wilson, D. W. N., Hahn, A. W., Weyhermüller, T., Van Stappen, C., Holland, P., DeBeer, S. (2023). Sulfur-Ligated [2Fe-2C] Clusters as Synthetic Model Systems for Nitrogenase. INORGANIC CHEMISTRY, 62(6), 2663-2671. doi:10.1021/acs.inorgchem.2c03693.
- Antico, E., Leutzsch, M., Wessel, N., Weyhermüller, T., Werlé, C., Leitner, W. (2022). Selective oxidation of silanes into silanols with water using [MnBr(CO)(5)] as a precatalyst. Chemical Science, 14(1), 54-60. doi:10.1039/d2sc05959b.
- Harihar, S., Mone, N., Satpute, S. K., Chadar, D., Chakravarty, D., Weyhermüller, T., Butcher, R. J., Salunke-Gawali, S. (2022). Metal complexes of a pro-vitamin K3 analog phthiocol (2-hydroxy-3-methylnaphthalene-1,4-dione): synthesis, characterization, and anticancer activity. Dalton Transactions, (51), 17338-17353. doi:10.1039/d2dt02748h.
- Chugh, V., Chatterjee, B., Chang, W.-C., Cramer, H. H., Hindemith, C., Randel, H., Weyhermüller, T., Fares, C., Werle, C. (2022). An Adaptive Rhodium Catalyst to Control the Hydrogenation Network of Nitroarenes. Angewandte Chemie, International Edition in English, (61): e202205515, pp. 1-10. doi:10.1002/anie.202205515.
- Henthorn, J. T., Cutsail III, G. E., Weyhermüller, T., DeBeer, S. (2022). Stabilization of intermediate spin states in mixed-valent diiron dichalcogenide complexes. Nature Chemistry, (14), 328-333. doi:10.1038/s41557-021-00853-5.
- Shit, M., Maity, S., Bera, S., Weyhermüller, T., Ghosh, P. (2022). Coordination of o-benzosemiquinonate, o-iminobenzosemiquinonate, 4, 4 '-di-tert-butyl-2,2 '-bipyridine and 1,10-phenanthroline anion radicals to oxidovanadium(IV) (vol 40, pg 10305, 2016). New Journal of Chemistry, 46(7), 3521-3521. doi:10.1039/d2nj90016e.
- Chang, W.-C., Deufel, F., Weyhermüller, T., Fares, C., Werlé, C. (2021). Rhodium(I) complexes derived from tris(isopropyl)-azaphosphatrane-controlling the metal-ligand interplay. RSC Advances, 11(59), 37383-37391. doi:10.1039/d1ra07126b.
- Kinzel, N. W., Demirbas, D., Bill, E., Weyhermüller, T., Werlé, C., Kaeffer, N., Leitner, W. (2021). Systematic Variation of 3d Metal Centers in a Redox-Innocent Ligand Environment: Structures, Electrochemical Properties, and Carbon Dioxide Activation. Inorganic Chemistry, (xx), xx-xx. doi:10.1021/acs.inorgchem.1c02909.
- Perdomenico, J., Levin, N., Fierro, A. C., Chernek, O. A. C., Weyhermüller, T., Slep, L. D. (2021). A New Member of the Growing Family of Interconvertible {RuNO}(6,7,8) Species. Redox and Acid-Base Characterization of [Ru((CH(2)py)(2)Me[9]aneN(3))(NO)](n+). European Journal of Inorganic Chemistry, 4842-4855. doi:10.1002/ejic.202100753.
- Chatterjee, B., Jena, S., Chugh, V., Weyhermüller, T., Werle, C. (2021). A Molecular Iron-Based System for Divergent Bond Activation: Controlling the Reactivity of Aldehydes. ACS CATALYSIS, 11(12), 7176-7185. doi:10.1021/acscatal.1c00733.
- Shit, M., Maity, S., Bera, S., Kumar Mudi, P., Biswas, B., Weyhermüller, T., Ghosh, P. (2021). Nickel (II) di-aqua complex containing water cluster: Synthesis, X-ray structure and catecholase activity New Journal of Chemistry, 45, 2221--2227 https://doi.org/10.1039/D0NJ05238H
- Voit, G., Jenthra, S., Hölscher, M., Weyhermüller, T., Leitner, W. (2020). Reversible Insertion of Carbon Dioxide at Phosphine Sulfonamido PdII–Aryl Complexes Organometallics 39(24), 4465-4473. https://doi.org/10.1021/acs.organomet.0c00560
- Levin, N., Peredkov, S., Weyhermüller, T., Rüdiger, O., Pereira, N.B., Grötzsch, D., Kalinko, A., DeBeer, S. (2020). Ruthenium 4d-to-2p X-ray Emission Spectroscopy: A Simultaneous Probe of the Metal and the Bound Ligands Inorganic Chemistry 59(12), 8272-8283. https://doi.org/10.1021/acs.inorgchem.0c00663
- Cramer, H.H., Chatterjee, B., Weyhermüller, T., Werlé, C., Leitner, W. (2020). Controlling the Product Platform of Carbon Dioxide Reduction: Adaptive Catalytic Hydrosilylation of CO2 Using a Molecular Cobalt(II) Triazine Complex Angewandte Chemie International Edition 59(36), 15674-15681. https://doi.org/10.1002/anie.202004463
- Erken, C., Hindemith, C., Weyhermüller, T., Hölscher, M., Werlé, C., Leitner, W. (2020). Hydroamination of Aromatic Alkynes to Imines Catalyzed by Pd(II)–Anthraphos Complexes ACS Omega 5(15), 8912-8918. https://doi.org/10.1021/acsomega.0c00562
- Dutta, D., Kundu, S., Weyhermüller, T., Ghosh, P. (2020). Metal promoted conversion of aromatic amines to ortho-phenylenediimine derivatives by a radical coupling path Dalton Transactions 49(16), 5015-5019. https://doi.org/10.1039/D0DT00089B
- Dinda, S., Patra, S.C., Roy, S., Halder, S., Weyhermüller, T., Pramanik, K., Ganguly, S. (2020). Coligand driven diverse organometallation in benzothiazolyl-hydrazone derivatized pyrene: ortho vs peri C–H activation New Journal of Chemistry 44(4), 1407-1417. https://doi.org/10.1039/c9nj05088d
- Yogendra, S., Weyhermüller, T., Hahn, A.W., DeBeer, S. (2019). From Ylides to Doubly Yldiide-Bridged Iron(II) High Spin Dimers via Self-Protolysis Inorganic Chemistry 58(14), 9358-9367. https://doi.org/10.1021/acs.inorgchem.9b01086
- Kalläne, S.I., Hahn, A.W., Weyhermüller, T., Bill, E., Neese, F., DeBeer, S., van Gastel, M. (2019). Spectroscopic and Quantum Chemical Investigation of Benzene-1,2- dithiolate-Coordinated Diiron Complexes with Relevance to Dinitrogen Activation Inorganic Chemistry 58(8), 5111-5125. https://doi.org/10.1021/acs.inorgchem.9b00177
- Bhand, S., Landem D.N., Pereira, E., Gejji, S.P., Weyhermüller, T., Chakravarty, D., Puranik, V.G., Salunke-Gawali, S. (2019). Amphiphilic polypyridyl ruthenium complexes: Synthesis, Characterization and Aggregation studies Polyhedron 164, 96-107. https://doi.org/10.1016/j.poly.2019.02.035
- Römelt, C., Weyhermüller, T., Wieghardt, K. (2019). Structural characteristics of redox-active pyridine-1,6-diimine complexes: Electronic structures and ligand oxidation levels Coordination Chemistry Reviews 380, 287-317. https://doi.org/10.1016/j.ccr.2018.09.018
- Wang, M., Römelt, C., Weyhermüller, T., Wieghardt, K. (2019). Coordination Modes, Oxidation, and Protonation Levels of 2,6-Pyridinediimine and 2,2′:6′,2′́-Terpyridine Ligands in New Complexes of Cobalt, Zirconium, and Ruthenium. An Experimental and Density Functional Theory Computational Study Inorganic Chemistry 58(1), 121-132. https://doi.org/10.1021/acs.inorgchem.8b01949
- Erken, C., Kaithal, A., Sen, S., Weyhermüller, T., Hölscher, M., Werlé, C., Leitner, W. (2018). Manganese-catalyzed hydroboration of carbon dioxide and other challenging carbonyl groups Nature Communications 9, 4521. https://doi.org/10.1038/s41467-018-06831-9
- Kundu, S., Dutta, D., Maity, S., Weyhermüller, T., Ghosh, P. (2018). Proton-Coupled Oxidation of a Diarylamine: Amido and Aminyl Radical Complexes of Ruthenium(II) Inorganic Chemistry 57(19), 11948-11960. https://doi.org/10.1021/acs.inorgchem.8b01401
- Levin, N., Codesido N.O., Marcolongo, J.P., Alborés, Weyhermüller, T., Olabe, J.A., Slep, L.D. (2018). Remarkable Changes of the Acidity of Bound Nitroxyl (HNO) in the [Ru(Me3[9]aneN3)(L2)(NO)]n+ Family (n = 1-3). Systematic Structural and Chemical Exploration and Bioinorganic Chemistry Implications Inorganic Chemistry 57(19), 12270-12281. https://doi.org/10.1021/acs.inorgchem.8b01958
- Hahn, A.W., Van Kuiken, B.E., Chilkuri, V.G., Levin, N., Bill, E., Weyhermüller, T., Nicolaou, A., Miyawaki, J., Harada, Y., DeBeer, S. (2018). Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X ray Scattering (RIXS) Inorganic Chemistry 57(37), 11918-11923. https://doi.org/10.1021/acs.inorgchem.8b01550
- Van Kuiken, B.E., Hahn, A.W., Nayyar, B., Schiewer, C.E., Lee, S.C., Meyer, F., Weyhermüller, T., Nicolaou, A., Cui, Y-T., Miyawaki, J., Hatada, Y., DeBeer, S. (2018). Electronic Spectra of Iron-Sulfur Complexes Measured by 2p3d RIXS Spectroscopy Inorganic Chemistry 57(12), 7355-7361. https://doi.org/10.1021/acs.inorgchem.8b01010
- Römelt, C., Ye, S., Bill, E., Weyhermüller, T., van Gastel, M., Neese, F. (2018). Electronic Structure and Spin Multiplicity of Iron Tetraphenylporphyrins in their Reduced States as Determined by a Combination of Resonance Raman Spectroscopy and Quantum Chemistry Inorganic Chemistry 57(4), 2141-2148. doi.org/10.1021/acs.inorgchem.7b03018
- Rees, J.A., Bjornsson, R., Kowalska, J.K., Lima, F.A., Schlesier, J., Sippel, D., Weyhermüller, T., Einsle, O., Kovacs, J.A., DeBeer, S. (2017). Comparative electronic structures of nitrogenase FeMoco and FeVco Dalton Transactions 46(8), 2445-2455. https://doi.org/10.1039/c7dt00128b
- Suturina, E.A., Nehrkorn, J., Zadrozny, J.M., Liu, J., Atanasov, M., Weyhermüller, T., Maganas, D., Hill, S., Schnegg, A., Bill, E., Long, J.R., Neese, F. (2017). Magneto-Structural Correlations in Pseudotetrahedral Forms of the [Co(SPh)4]2- Complex Probed by Magnetometry, MCD Spectroscopy, Advanced EPR Techniques, and ab Initio Electronic Structure Calculations Inorganic Chemistry 56(5), 3102-3118. https://doi.org/10.1021/acs.inorgchem.7b00097
- Chowdhury, A.R., Roy, B.G., Jana, S., Weyhermüller, T., Banerjee, P. (2017). A simple cleft shaped hydrazine-functionalized colorimetric new Schiff base chemoreceptor for selective detection of F- in organic solvent through PET signaling: Development of a chemoreceptor based sensor kit for detection of fluoride Sensors and Actuators B: Chemical 241, 706-715. https://doi.org/10.1016/j.snb.2016.10.095
- Römelt, C., Song, J.S., Tarrago, M., Rees, J.A., van Gastel, M., Weyhermüller, T., DeBeer, S., Bill, E., Neese, F., Ye, S. (2017). Electronic Structure of a Formal Iron(0),Porphyrin Complex Relevant to CO2 Reduction Inorganic Chemistry 56(8), 4745-4750. https://doi.org/10.1021/acs.inorgchem.7b00401
- Manikandamathavan V.M., Thangaraj M., Weyhermüller T., Parameswari R.P., Punitha V., Murthy N.N., Nair B.U. (2017). Novel mononuclear Cu(II) terpyridine complexes: Impact of fused ring thiophene and thiazole head groups towards DNA/BSA interaction, cleavage and antiproliferative activity on HepG2 and triple negative CAL-51 cell line European Journal of Medicinal Chemistry 135(28), 434-446. https://doi.org/10.1016/j.ejmech.2017.04.030
- Sabenya, G., Lázaro, L., Gambo, I., Martin-Diaconescu, V., Andris, E., Weyhermüller, T., Neese, F., Roithova, J., Bill, E., Lloret-Fillol, J., Costas, M. (2017). Generation, Spectroscopic, and Chemical Characterization of an Octahedral Iron(V)-Nitrido Species with a Neutral Ligand Platform Journal of the American Chemical Society 139(27), 9168-9177. https://doi.org/10.1021/jacs.7b00429
- Hahn, A.W., Van Kuiken, B.E., al Samarai, M., Atanasov, M., Weyhermüller, T., Cui, Y.T., Miyawaki, J., Harada, Y., Nicolaou, A., DeBeer, S. (2017). Measurement of the Ligand Field Spectra of Ferrous and Ferric Iron Chlorides Using 2p3d RIXS Inorganic Chemistry 56(14), 8203-8211. https://doi.org/10.1021/acs.inorgchem.7b00940
- Kowalska, J.K., Nayyar, B., Rees, J.A., Schiewer, C.E., Lee, S.C., Kovacs, J.A., Meyer, F., Weyhermüller, T., Otero, E., DeBeer, S. (2017). Iron L2,3-edge X-ray Absorption and X-ray Magnetic Circular Dichroism Studies of Molecular Iron Complexes with Relevance to the FeMoco and FeVco Active Sites of Nitrogenase Inorganic Chemistry 56(14), 8147-8158. https://doi.org/10.1021/acs.inorgchem.7b00852
- Khannam, M., Weyhermüller, T., Goswami, U., Mukherjee, C. (2017). A highly stable L-alanine-based mono(aquated) Mn(II) complex as a T1-weighted MRI contrast agent Dalton Transactions 46(31), 10426-10432. https://doi.org/10.1039/c7dt02282d
- Shit, M., Bera, S., Maity, S., Weyhermüller, T., Ghosh, P. (2017). Coordination of o-benzosemiquinonate, o-iminobenzosemiquinonate and aldimine anion radicals to oxidovanadium(IV) New Journal of Chemistry 41, 4564-4572. https://doi.org/10.1039/c7nj00186j
- Levin, N. Perdoménico, J., Bill, E., Weyhermüller, T., Slep, L.D. (2017). Pushing the photodelivery of nitric oxide to the visible: are {FeNO}7 complexes good candidates? Dalton Transactions 46(46), 16058-16064. https://doi.org/10.1039/c7dt03142d
- Shit, M., Bera, S., Maity, S., Maji, S., Weyhermüller, T., Ghosh, P. (2016). Oxidovanadium Complexes of 2,2'-Bipyridine, 1,10 Phenanthroline, and p-Nitro-o-aminophenol - Radical versus Nonradical States European Journal of Inorganic Chemistry 2016(3), 330-338. https://doi.org/10.1002/ejic.201501246
- Bera, S., Maity, S., Weyhermüller, T., Ghosh, P. (2016). Radical non-radical states of the [Ru(PIQ)] core in complexes (PIQ=9,10-phenanthreneiminoquinone) Dalton Transactions 45(19), 8236-8247. https://doi.org/10.1039/c6dt00091f
- Bera, S., Mondal, S., Maity, S., Weyhermüller, T., Ghosh, P. (2016). Radical and Non-Radical States of the [Os(PIQ)] Core (PIQ = 9,10-Phenanthreneiminoquinone): Iminosemiquinone to Iminoquinone Conversion Promoted o-Metalation Reaction Inorganic Chemistry 55(10), 4746-4756. https://doi.org/10.1021/acs.inorgchem.6b00040
- Wang, M., Weyhermüller, T., Bill, E., Ye, S., Wieghardt, K. (2016). Structural and Spectroscopic Characterization of Rhenium Complexes Containing Neutral, Monoanionic, and Dianionic Ligands of 2,2'-Bipyridines and 2,2':6,2''-Terpyridines: An Experimental and Density Functional Theory (DFT)-Computational Study Inorganic Chemistry 55(10), 5019-5036. https://doi.org/10.1021/acs.inorgchem.6b00609
- Dar, U.A., Shand, S., Lande, D.N., Rao, S.S., Patil, Y.P., Gejji, S.P., Nethaji, M., Weyhermüller, T., Salunke-Gawali, S. (2016). Molecular structures of 2-hydroxy-1,4-naphthoqinone derivatives and their zinc(II) complexes: Combining experiment and density functional theory Polyhedron 113, 61 72. https://doi.org/10.1016/j.poly.2016.04.002
- Bhand, S., Patil, R., Shinde, Y., Lande, D.N., Rao, S.S., Kathawate, L., Gejji, S.P., Weyhermüller, T., Salunke-Gawali, S. (2016). Tautomerism in o-hydroxyanilino-1,4-naphthoquinone derivatives: Structure, NMR, HPLC and density functional theoretic investigations Journal of Molecular Structure 1123, 245-260. https://doi.org/10.1016/j.molstruc.2016.06.026
- Kundu, S., Mondal, A., Weyhermüller, T., Sproules, S., Ghosh, P. (2016). Molecular and electronic structures of copper-cuprizone and analogues Inorganica Chimica Acta 451, 23-30. https://doi.org/10.1016/j.ica.2016.06.040
- Maity, S., Kundu, S., Bera, S., Weyhermüller, T., Ghosh, P. (2016). Mixed-Valence o-Iminobenzoquinone and o-Iminobenzosemiquinonate Anion Radical Complexes of Cobalt: Valence Tautomerism European Journal of Inorganic Chemistry 2016(22), 3680-3690. https://doi.org/10.1002/ejic.201600525
- Maity, S., Kundu, S., Bera, S., Weyhermüller, T., Ghosh, P. (2016). o-Iminobenzoquinone and o-Iminobenzosemiquinonate Anion Radical Complexes of Rhodium and Ruthenium European Journal of Inorganic Chemistry 2016(22), 3691-3697. https://doi.org/10.1002/ejic.201600526
- Levin, N., Codesido, N.O., Bill, E., Weyhermüller, T., Gaspari, A.P.S., da Silva, R.S., Olabe, J.A., Slep, L.D. (2016). Structural, Spectroscopic, and Photochemical Investigation of an Octahedral NO Releasing {RuNO}7 Species Inorganic Chemistry 55(16), 7808-7810. https://doi.org/10.1021/acs.inorgchem.6b00719
- Shit, M., Maity, S., Bera, S., Weyhermüller, T., Ghosh, P. (2016). Coordination of o-benzosemiquinonate, o-iminobenzosemiquinonate, 4,4'-di-tert-butyl-2,2'-bipyridine and 1,10-phenanthroline anion radicals to oxidovanadium(IV) New Journal of Chemistry 40(12), 10305-10315. https://doi.org/10.1039/c6nj02220k
- Bera, S., Maity, S., Weyhermüller, T., Ghosh, P. (2016). Arylamino radical complexes of ruthenium and osmium: dual radical counter in a molecule Dalton Transactions 45(48), 19428-19440. https://doi.org/10.1039/c6dt03728c
Chemische Synthese, Röntgenstrukturanalyse, NMR
Synthese von Modellverbindungen
Die Forschung in meiner Gruppe konzentriert sich auf die Synthese kleiner molekularer Modellkomplexe für spektroskopische Untersuchungen. Systematische Variationen von strukturellen oder elektronischen Merkmalen in einer Serie von Verbindungen mit sonst gleichem Aufbau erlauben es, die beobachteten spektroskopischen Änderungen mit der Struktur zu korrelieren. Unsere Verbindungen werden mittels Standardmethoden analysiert und die Strukturen bestimmt, bevor sie weiter spektroskopisch untersucht werden (Mössbauer, EPR, SQUID, XAS, etc.). Modellverbindungen deren Struktur und Zusammensetzung sehr genau bekannt ist, ermöglichen es verlässliche und hochaufgelöste spektroskopische Daten zu sammeln die es aussderdem erlauben quantenchemische Verfahren zur Berechnung von spektroskopischen Parametern zu überprüfen und zu kalibrieren.
Modellsysteme für die Nitrogenase
Wir beschäftigen uns seit einiger Zeit mit der Modellierung des katalytisch aktiven Zentrums der Nitrogenase. Es handelt sich dabei um ein bakteielles Enzym das die Umwandlung von Luftstickstoff zu NH3 katalysiert. Dieses Ammoniak ist wiederum die primäre Quelle für die Biosynthese von essentiellen stickstoffhaltigen Verbindungen in diesen Bakterien und von den mit ihnen in Symbiose lebenden Pflanzen.
Die chemische Aktivierung von Stickstoff stellt eine enorme Herausforderung dar, da das Stickstoffmolekül mit seiner Dreifachbindung das reaktionsträgste zweiatomige Molekül überhaupt ist. Mittlerweile ist über Aufbau und Chemie der Nitrogenase sehr viel bekannt, leider liegen die genaue elektronische Struktur im Reaktionszyklus, der katalytische Mechanismus, die Funktion des zentralen Kohlenstoffatoms [1], oder des Molydänatoms im FeMoco Faktor nach wie vor im Dunkeln. Der metallhaltige Kofaktor FeMoco wurde als katalytisch aktives Zentrum des Enzyms identifiziert an dem der Stickstoff gebunden und zu Ammoniak umgesetzt wird. Er besteht im Wesentlichen aus sieben Eisen-, einem Molybdänzentrum, neun Sulfidionen und einem zentralen Carbidion (s. Abb. 1 unten rechts). Wir sind besonders an der chemischen Funktion des Carbidions im Cluster interessiert und haben deshalb versucht Modellsysteme zu synthetisieren die Ähnlichkeiten zum natürlichen Vorbild aufweisen.
Wir haben eine Serie von kohlenstoffverbrückten zweikernigen Eisenkomplexen untersucht, die durch Reaktion von Bis(diphenylthiophosphinoyl)methanediide2- mit FeCl2 in THF zugänglich sind [2]. Abb. 1 zeigt den Aufbau und die Struktur der Verbindung 1 und macht die Ähnlichkeit zur Struktur von FeMoco (untere Zeile) deutlich.
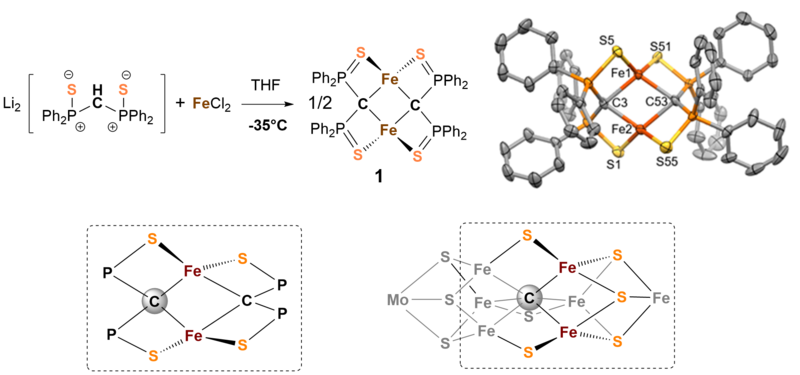
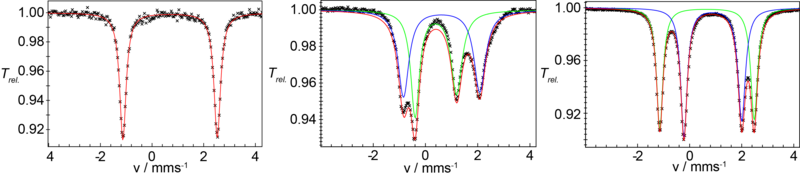
Verbindung 1 kann elektrochemisch reversibel oder mit einem geeigneten Oxiationsmittel chemisch sauber in den gemischtvalenten Fe(II)Fe(III)-Komplex 2 überführt werden. Abb. 2 zeigt die 57Fe-Mössbauerspektren von 1 und 2. Die Spektren zeigen eindeutig, dass die Fe(II) und Fe(III) Zentren in beiden Komplexen high-spin Elektronenkonfiguration besitzen.
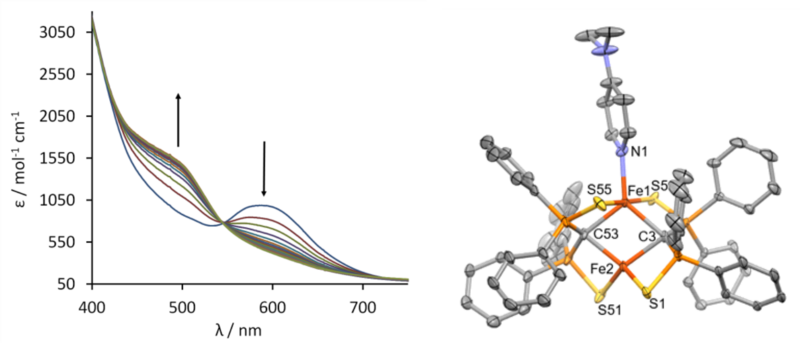
Verbindung 1 kann auch geeignte Substrate unter Aufweitung der Koordinationssphäre am Eisen binden. Abb. 3 zeigt die Änderung des UV/vis Spektrums in Abbhängigkeit der zudosierten Menge an Substrat, in diesem Falle Dimethylaminopyridin (DMAP). Die spektrometrische Titration und auch die Röntgenstrukturanalyse des Produkts zeigen, dass 1 genau ein Molekül DMAP unter Bildung von 3 addiert (s. Abb 3)
Strukturaufklärung mittels Röntgenbeugung und NMR-Spektroskopie
Die Kenntnis über den genauen dreidimensionalen Aufbau der Modellverbindungen und katalytisch aktiven Verbindungen die in unseren Abteilungen untersucht werden ist von großer Bedeutung um die Eigenschaften und die Reaktivität dieser Moleküle zu verstehen. Die ermittelte Struktur der Metallkomplexe bildet auch die Grundlage quantenmechanischer Rechnungen an diesen Substanzen, die wiederum dem Verständnis von Reaktivität und spektroskopischen Eigenschaften dienen die in der Abteilung „Anorganische Spektroskopie“ und dem „Joint Workspace“ untersucht werden.
Die Arbeit in meiner Gruppe konzentriert sich auf die Strukturaufklärung von kleinen und mittelgroßen Molekülen mittels Röntgendiffraktometrie und NMR-Spektroskopie.
Forschung an molekularen Übergangsmetallverbindungen für die Katalyse ist auf die Strukturbestimmung mittels Röntgenbeugung an Einkristallen angewiesen, da Selbstorganisationseffekte die Koordinationschemie oft dominieren und die gezielte Synthese von Verbindungen nur eingeschränkt möglich ist. Die Aufklärung der Struktur mittels anderer Methoden, wie der NMR-Spektroskopie, ist oft nicht möglich, da bei Anwesenheit von paramagnetischen Metallzentren normalerweise keine hochaufgelösten Spektren erhalten werden können. Die Röntgenstrukturanalyse am Einkristall liefert davon unabhängig hochpräzise Informationen über die dreidimensionale Anordnung der Atome und produziert genaue Bindungslängen und Bindungswinkel, die von grundlegender Bedeutung für das Verständnis der chemischen Eigenschaften einer Verbindung sind. Voaraussetzung für die Ermittlung der Struktur mittels dieser Methode sind allerdings Einkristalle die nicht immer einfach, manchmal gar nicht zu erhalten sind.
Für die Strukturbestimmung in Lösung ist die NMR-Spektroskopie sehr gut geeignet. Sie liefert bei diamagnetischen Verbindungen detaillierte Informationen über die Konnektivität von Atomen in Molekülen, kann aber keine genauen Werte für Bindungslängen und Winkeln ermitteln. Insbesondere zur schnellen Beurteilung von synthetischen Zwischenprodukten und zur Charakterisierung von Zielverbindungen ist sie aber ein hervorragendes und unverzichtbares Mittel.


Referenzen
[1] a) Lancaster, K. M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M. W.; Neese, F.; Bergmann, U.; DeBeer, S., X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334 (6058), 974. b) Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S. L. A.; Schleicher, E.; Weber, S.; Rees, D. C.; Einsle, O., Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334 (6058), 940-940.
[2] a) Yogendra, S.; Wilson, D.; Hahn, A.; Weyhermüller, T.; van Stappen, C.; Holland, P.; DeBeer, S., Sulfur-Ligated [2Fe-2C]-Clusters as Synthetic Model Systems for Nitrogenase subm. Inorg. Chem. 2022 under review b) Fustier-Boutignon, M.; Heuclin, H.; Le Goff, X. F.; Mezailles, N., Transmetalation of a nucleophilic carbene fragment: from early to late transition metals. Chem. Commun., 2012, 48 (27), 3306-8.