Prof. Dr. Serena DeBeer - Anorganische Spektroskopie

- Prof. Dr. Serena DeBeer
- Direktorin
- Anorganische Spektroskopie
- +49 (0)208 306 - 3656
- serena.debeer(at)cec.mpg.de
- Raum: 608
Vita
| B.S. (Chemie) | Southwestern University, TX, USA (1995) |
| Ph.D. (Chemie) | Stanford University (2002) |
| Beam Line Scientist | SSRL, SLAC, Stanford, University (2001-2003) |
| Staff Scientist | SSRL, SLAC, Stanford, University (2003-2009) |
| Assistenz-Professor | Cornell University, NY, USA (2009-2012) |
| Forschungsgruppenleiterin | MPI für Bioanorganische Chemie; heute: MPI CEC (2011-2017) |
| Gruppenleiterin | PINK Beamline, Energy Materials In-Situ Laboratory, Helmholtz Zentrum, Berlin (seit 2012) |
| Adjunct Associate Professor | Department of Chemistry and Chemical Biology, Cornell University, NY, USA (seit 2012) |
| Honorarprofessur | Ruhr Universität Bochum (seit 2014) |
| Direktorin | Anorganische Spektroskopie, MPI CEC (seit 2017) |
| Honorarprofessur | Universität Duisburg-Essen (seit 2024) |
Publications
Full publications list | ORCID | ResearcherID | Google Scholar Profile
MPI CEC publications
- Chrysina, M., Drosou, M., Castillo, R. G., Reus, M., Neese, F., Krewald, V., Pantazis, D. A., DeBeer, S. (2023). Nature of S-States in the Oxygen-Evolving Complex Resolved by High-Energy Resolution Fluorescence Detected X-ray Absorption Spectroscopy. Journal of the American Chemical Society, 145(47), 25579-25594. doi:10.1021/jacs.3c06046.
- Sodreau, A., Zahedi, H. G., Dervisoglu, R., Kang, L., Menten, J., Zenner, J., Terefenko, N., DeBeer, S., Wiegand, T., Bordet, A., Leitner, W. (2023). A Simple and Versatile Approach for the Low-Temperature Synthesis of Transition Metal Phosphide Nanoparticles from Metal Chloride Complexes and P(SiMe3)3. ADVANCED MATERIALS. 2306621 (1-9) doi:10.1002/adma.202306621.
- Wandzilak, A., Grubel, K., Skubi, K. L., McWilliams, S. F., Bessas, D., Rana, A., Hugenbruch, S., Dey, A., Holland, P. L., DeBeer, S. (2023). Mössbauer and Nuclear Resonance Vibrational Spectroscopy Studies of Iron Species Involved in N-N Bond Cleavage. INORGANIC CHEMISTRY, 62(45), 18449-18464. doi:10.1021/acs.inorgchem.3c02594.
- DeBeer, S., Moonshiram, D. (2023). Mapping the Ultrafast Mechanistic Pathways of Co Photocatalysts in Pure Water through Time-Resolved X-ray Spectroscopy. ChemSusChem, e202300719, pp. 1-14. doi:10.1002/cssc.202300719.
- Pang, Y., Nöthling, N., Leutzsch, M., Kang, L., Bill, E., van Gastel, M., Reijerse, E., Goddard, R., Wagner, L., SantaLucia, D., DeBeer, S., Neese, F., Cornella, J. (2023). Synthesis and isolation of a triplet bismuthinidene with a quenched magnetic response. Science, 380 (6649), 1043-1048. doi:10.1126/science.adg2833.
- Genoux, A., Pauly, M., Rooney, C. L., Choi, C., Shang, B., McGuigan, S., Fataftah, M. S., Kayser, Y., Suhr, S. C. B., DeBeer, S., Wang, H., Maggard, P. A., Holland, P. L. (2023). Well-Defined Iron Sites in Crystalline Carbon Nitride. JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 145(38), 20739-20744. doi:10.1021/jacs.3c05417.
- Zhang, Y., El Sayed, S., Kang, L., Sanger, M., Wiegand, T., Jessop, P. G., DeBeer, S., Bordet, A., Leitner, W. (2023). Adaptive Catalysts for the Selective Hydrogenation of Bicyclic Heteroaromatics using Ruthenium Nanoparticles on a CO2-Responsive Support. Angewandte Chemie, International Edition in English, (XX): e202311427, pp. x-xx. doi:10.1002/anie.202311427.
- Hau, J.-L., Kaltwasser, S., Muras, V., Casutt, M. S., Vohl, G., Claussen, B., Steffen, W., Leitner, A., Bill, E., Cutsail, G. E., DeBeer, S., Vonck, J., Steuber, J., Fritz, G. (2023). Conformational coupling of redox-driven Na+-translocation in <i>Vibrio cholerae</i> NADH:quinone oxidoreductase. Nature Structural & Molecular Biology, (xx), 1-31. doi:10.1038/s41594-023-01099-0.
- Erbe, A., Tesch, M. F., Rüdiger, O., Kaiser, B., DeBeer, S., Rabe, M. (2023). Operando studies of Mn oxide based electrocatalysts for the oxygen evolution reaction. Physical Chemistry Chemical Physics, (40), 26933-27894. doi:10.1039/d3cp02384b.
- Liu, Y., Chatterjee, S., Cutsail III, G. E., Peredkov, S., Gupta, S. K., Dechert, S., DeBeer, S., Meyer, F. (2023). Cu4S Cluster in "0-Hole" and "1-Hole" States: Geometric and Electronic Structure Variations for the Active Cu-Z* Site of N2O Reductase. Journal of the American Chemical Society, 145(33), 18477-18486. doi:10.1021/jacs.3c04893.
- Hall, K. R., Joseph, C., Ayuso-Fernandez, I., Tamhankar, A. V., Rieder, L., Skaali, R., Golten, O., Neese, F., Rohr, A. K., Venturinelli Jannuzzi, S. A., DeBeer, S., Eijsink, V. G. H., Sorlie, M. (2023). A Conserved Second Sphere Residue Tunes Copper Site Reactivity in Lytic Polysaccharide Monooxygenases. Journal of the American Chemical Society, 145(34), 18888-18903. doi:10.1021/jacs.3c05342.
- Mao, W., Zhang, Z., Fehn, D., Venturinelli Jannuzzi, S. A., Heinemann, F., Scheurer, A., van Gastel, M., DeBeer, S., Munz, D., Meyer, K. (2023). Synthesis and Reactivity of a Cobalt-Supported Singlet Nitrene. Journal of the American Chemical Society, 145(25), 13650-13662. doi:10.1021/jacs.3c01478.
- Pielsticker, L., Nicholls, R. L., DeBeer, S., Greiner, M. (2023). Convolutional neural network framework for the automated analysis of transition metal X-ray photoelectron spectra. Analytica Chimica Acta, (1271) 341433 doi:10.1016/j.aca.2023.341433.
- Van Stappen, C., Benediktsson, B., Rana, A., Chumakov, A., Yoda, Y., Bessas, D., Decamps, L. B., Bjornsson, R., DeBeer, S. (2023). Structural correlations of nitrogenase active sites using nuclear resonance vibrational spectroscopy and QM/MM calculations. Faraday Discussions, (243) 253-269.. doi:10.1039/d2fd00174h.
- Kovel, C. B., Darmon, J. M., Stieber, S. C. E., Pombar, G., Pabst, T. P., Theis, B., Turner, Z. R., Ungör, Ö., Shatruk, M., DeBeer, S., Chirik, P. J. (2023). Bimolecular Reductive Elimination of Ethane from Pyridine(diimine) Iron Methyl Complexes: Mechanism, Electronic Structure, and Entry into [2+2] Cycloaddition Catalysis. Journal of the American Chemical Society, (145), 5061-5073. doi:10.1021/jacs.2c10547.
- Martini, M. A., Bikbaev, K., Pang, Y., Lorent, C., Wiemann, C., Breuer, N., Zebger, I., DeBeer, S., Span, I., Bjornsson, R., Birrell, J. A., Rodriguez-Macia, P. (2023). Binding of exogenous cyanide reveals new active-site states in [FeFe] hydrogenases. Chemical Science, (14) 2826-2838. doi:10.1039/d2sc06098a.
- Yogendra, S., Wilson, D. W. N., Hahn, A. W., Weyhermüller, T., Van Stappen, C., Holland, P., DeBeer, S. (2023). Sulfur-Ligated [2Fe-2C] Clusters as Synthetic Model Systems for Nitrogenase. INORGANIC CHEMISTRY, 62(6), 2663-2671. doi:10.1021/acs.inorgchem.2c03693.
- Anandaraj, S. J. L., Kang, L., DeBeer, S., Bordet, A., Leitner, W. (2023). Catalytic Hydrogenation of CO2 to Formate Using Ruthenium Nanoparticles Immobilized on Supported Ionic Liquid Phases. Small,(19) 2206806, pp. 1-10. doi:10.1002/smll.202206806.
- Caserta, G., Hartmann, S., Van Stappen, C., Karafoulidi-Retsou, C., Lorent, C., Yelin, S., Keck, M., Schoknecht, J., Sergueev, I., Yoda, Y., Hildebrandt, P., Limberg, C., DeBeer, S., Zebger, I., Frielingsdorf, S., Lenz, O. (2023). Stepwise assembly of the active site of [NiFe]-hydrogenase. Nature Chemical Biology,(19) 498-506. doi:10.1038/s41589-022-01226-w.
- Cutsail III, G., Schott-Verdugo, S., Müller, L., DeBeer, S., Groth, G., Gohlke, H. (2022). Spectroscopic and QM/MM studies of the Cu(I) binding site of the plant ethylene receptor ETR1. BIOPHYSICAL JOURNAL, 121(20), 3862-3873. doi:10.1016/j.bpj.2022.09.007.
- Keilwerth, M., Mao, W., Venturinelli Jannuzzi, S. A., Grunwald, L., Heinemann, F. W., Scheurer, A., Sutter, J., De Beer, S., Munz, D.,Meyer, K., (2023). From Divalent to Pentavalent Iron Imido Complexes and an Fe(V) Nitride via N-C Bond Cleavage. Journal of the American Chemical Society, (145), 873-887. doi:10.1021/jacs.2c09072.
- Bowker, M., DeBeer, S., Dummer, N. F., Hutchings, G. J., Scheffler, M., Schüth, F., Taylor, S., Tueysuez,H. (2022). Advancing Critical Chemical Processes for a Sustainable Future: Challenges for Industry and the Max Planck-Cardiff Centre on the Fundamentals of Heterogeneous Catalysis (FUNCAT). Angewandte Chemie, International Edition in English, (61): e202209016, pp. 1-13. doi:10.1002/anie.202209016.
- Alkan, B., Braun, M., Landrot, G., Rüdiger, O., Andronescu, C., DeBeer, S., Schulz, C., Wiggers, H. (2022). Spray-flame-synthesized Sr- and Fe-substituted LaCoO3 perovskite nanoparticles with enhanced OER activities. Journal of Materials Science, (57), 18923-18936. doi:10.1007/s10853-022-07738-z.
- Yu, M., Weidenthaler, C., Wang, Y., Budiyanto, E., Sahin, E. O., Chen, M., DeBeer, S., Rüdiger, O., Tueysuez, H. (2022). Surface Boron Modulation on Cobalt Oxide Nanocrystals for Electrochemical Oxygen Evolution Reaction. Angewandte Chemie, International Edition in English, (61): e202211543, pp. 1-12. doi:10.1002/anie.202211543.
- Mao, W., Fehn, D., Heinemann, F. W., Scheurer, A., van Gastel, M., Jannuzzi V, S. A., DeBeer, S., Munz, D., Meyer, K. (2022). Umpolung in a Pair of Cobalt(III) Terminal Imido/Imidyl Complexes. Angewandte Chemie International Edition, (61): e202206848, pp. 1-9. doi:10.1002/anie.202206848.
- Cutsail III, G. E., Banerjee, R., Rice, D. B., Stepanic, O. M., Lipscomb, J. D., DeBeer, S. (2022). Determination of the iron(IV) local spin states of the Q intermediate of soluble methane monooxygenase by K beta X-ray emission spectroscopy. Journal of Biological Inorganic Chemistry, (27), 573-582. doi:10.1007/s00775-022-01953-4.
- Decamps, L. B., Rice, D. B., DeBeer, S. (2022). An Fe6C Core in All Nitrogenase Cofactors. Angewandte Chemie International Edition (61) e202209190, pp. 1-3. doi:10.1002/anie.202209190.
- Cutsail III, G. E., DeBeer, S. (2022). Challenges and Opportunities for Applications of Advanced X-ray Spectroscopy in Catalysis Research. ACS Catalysis, 12(10), 5864-5886. doi:10.1021/acscatal.2c01016.
- Xiang, W., Yang, N., Li, X., Linnemann, J., Hagemann, U., Ruediger, O.,Heidelmann,M.; Falk, T., Aramani, M., DeBeer, S., Muhler, M.,Tschulik, K., Li, T.. (2022). 3D atomic-scale imaging of mixed Co-Fe spinel oxide nanoparticles during oxygen evolution reaction. Nature Communications, 13(179), 1-14. doi:10.1038/s41467-021-277.
- Aldous, L., Comba, P., DeBeer, S., Dey, A., Draksharapu, A., Duboc, C., Itoh, S., Karlin, K., Kundu, S., Lopez Domene, R., Marechal, J.-D., Mazumdar, S., Mukherjee, R., Parker, D., Pordea, A., Que, L., Rath, S. P., Sadler, P., Sastri, C., Schindler, S., Schunemann, V., Sen Gupta, S., Solomon, E. I., P. Stack, T. D. (2022). Small molecule activation and synthetic analogues: general discussion. Faraday Discussions, (234), 129-142. doi:10.1039/d2fd90012b.
- Anilkumar, A., Ash, P., Chakravarty, A. R., Comba, P., DeBeer, S., Dey, A., Draksharapu, A., Goswami, D., Itoh, S., Karlin, K., Lakshmi, K. V., Mazumdar, S., Pantazis, D., Parker, D., Que, L., Rajaraman, G., Rath, S. P., Sastri, C., Sen Gupta, S., Solomon, E. I. (2022). Electron transfer, spectroscopy and theory: general discussion. Faraday Discussions, 234(0), 245-263. doi:10.1039/d2fd90013k.
- Chatterjee, S., Harden, I., Bistoni, G., Castillo, R. G., Chabbra, S., van Gastel, M., Schnegg, A., Bill, E., Birrell, J. A., Morandi, B., Neese, F., DeBeer, S. (2022). A Combined Spectroscopic and Computational Study on the Mechanism of Iron-Catalyzed Aminofunctionalization of Olefins Using Hydroxylamine Derived N-O Reagent as the "Amino" Source and "Oxidant". Journal of the American Chemical Society, 144(6), 2637-2656. doi:10.1021/jacs.1c11083.
- Souilah, C., Venturinelli Jannuzzi, S. A., Demirbas, D., Ivlev, S., Swart, M., DeBeer, S., Casitas, A. (2022). Synthesis of Fe-III and Fe-IV Cyanide Complexes Using Hypervalent Iodine Reagents as Cyano-Transfer One-Electron Oxidants. Angewandte Chemie, International Edition in English, (61): e202201699, pp. 1-7. doi:10.1002/anie.202201699.
- Sisodiya-Amrute, S., Van Stappen, C., Rengshausen, S., Han, C., Sodreau, A., Weidenthaler, C., Tricard, S., DeBeer, S., Chaudret, B., Bordet, A., Leitner, W. (2022). <p>Bimetallic MxRu100_x nanoparticles (M = Fe, Co) on supported ionic liquid phases (MxRu100-x@SILP) as hydrogenation catalysts: Influence of M and M:Ru ratio on activity and selectivity</p>. Journal of Catalysis,(407), 141-148. doi:10.1016/j.jcat.2022.01.030.
- Czastka, K., Alsheikh Oughli, A., Rüdiger, O. ,DeBeer, S. (2022). Enzymatic X-ray Absorption Spectroelectrochemistry. Faraday Discussions, (234), 214-231. doi:10.1039/D1FD00079A.
- Henthorn, J. T., DeBeer, S. (2022). Selenium Valence-to-Core X-ray Emission Spectroscopy and K beta HERFD X-ray Absorption Spectroscopy as Complementary Probes of Chemical and Electronic Structure. Inorganic Chemistry, 61(6), 2760-2767. doi:10.1021/acs.inorgchem.1c02802.
- Van Stappen, C., Jimenez-Vicente, E., Perez-Gonzalez, A., Yang, Z.-Y., Seefeldt, L. C., DeBeer, S., Dean, D. R., Decamps, L. B. (2022). A conformational role for NifW in the maturation of molybdenum nitrogenase P-cluster. Chemical Science, 13(13), 3489-3500. doi:10.1039/d1sc06418e.
- Jedrzkiewicz, D., Mai, J., Langer, J., Mathe, Z., Patel, N., DeBeer, S., Harder, S. (2022). Access to a Labile Monomeric Magnesium Radical by Ball-Milling. Angewandte Chemie, International Edition in English, (xx): e202200511, pp. 1-7. doi:10.1002/anie.202200511.
- Levin, N., Casadevall, C., Cutsail III, G. E., Lloret-Fillol, J., DeBeer, S., Rüdiger, O. (2022). XAS and EPR in Situ Observation of Ru(V) Oxo Intermediate in a Ru Water Oxidation Complex**. CHEMELECTROCHEM, (9): e202101271, pp. 1-4. doi:10.1002/celc.202101271.
- Henthorn, J. T., Cutsail III, G. E., Weyhermüller, T., DeBeer, S. (2022). Stabilization of intermediate spin states in mixed-valent diiron dichalcogenide complexes. Nature Chemistry, (14), 328-333. doi:10.1038/s41557-021-00853-5.
- Geoghegan, B. L., Liu, Y., Peredkov, S., Dechert, S., Meyer, F., DeBeer, S., Cutsail III, G. E. (2022). Combining Valence-to-Core X-ray Emission and Cu K-edge X-ray Absorption Spectroscopies to Experimentally Assess Oxidation State in Organometallic Cu(I)/(II)/(III) Complexes. Journal of the American Chemical Society, 144(6), 2520-2534. doi:10.1021/jacs.1c09505.
- Czastka, K., Alsheikh Oughli, A., Rüdiger, O., DeBeer, S. (2021). Enzymatic X-ray Absorption Spectroelectrochemistry. Faraday Discussions, (xx), 1-14. doi:10.1039/x0xx00000x.
- Gerz, I., Venturinelli Jannuzzi, S. A., Hylland, K. T., Negri, C., Wragg, D. S., Öien-Ödegaard, S.,Tilset, M., Olybye, U., DeBeer, S., Amedjkouh, M. (2021). Structural Elucidation, Aggregation, and Dynamic Behaviour of N,N,N,N-Copper(I) Schiff Base Complexes in Solid and in Solution: A Combined NMR, X-ray Spectroscopic and Crystallographic Investigation. European Journal of Inorganic Chemistry. doi:10.1002/ejic.202100982.
- Spiller, N. B., Bjornsson, R., DeBeer, S., Neese, F. (2021). Carbon Monoxide Binding to the Iron–Molybdenum Cofactor of Nitrogenase: a Detailed Quantum Mechanics/Molecular Mechanics Investigation. Inorganic Chemistry, 60(23), 18031-18047. doi:10.1021/acs.inorgchem.1c02649.
- Martini, M. A., Rüdiger, O., Breuer, N., Nöring, B., DeBeer, S., Rodriguez-Macia, P., Birrell, J. (2021). The Nonphysiological Reductant Sodium Dithionite and [FeFe] Hydrogenase: Influence on the Enzyme Mechanism. Journal of the American Chemical Society, (xx), xx-xx. doi:10.1021/jacs.1c07322.
- Budiyanto, E., Zerebecki, S., Weidenthaler, C., Kox, T., Kenmoe, S., Spohr, E., DeBeer, S., Rüdiger, O., Reichenberger, S., Barcikowski, S., Tuysuz, H., (2021). Impact of Single-Pulse, Low-Intensity Laser Post-Processing on Structure and Activity of Mesostructured Cobalt Oxide for the Oxygen Evolution Reaction. ACS applied materials & interfaces. doi:10.1021/acsami.1c08034.
- Gil-Sepulcre, M., Lindner, J. O., Schindler, D., Velasco, L., Moonshiram, D., Rüdiger, O., De Beer, S., Stepanenko, V., Solano, E., Würthner, F., Llobet, A. (2021). Surface-Promoted Evolution of Ru-bda Coordination Oligomers Boosts the Efficiency of Water Oxidation Molecular Anodes. Journal of the American Chemical Society, 143(30), 11651-11661. doi:10.1021/jacs.1c04738.
- Wang, C.-H., DeBeer, S. (2021) Structure, reactivity, and spectroscopy of nitrogenase-related synthetic and biological clusters. CHEMICAL SOCIETY REVIEWS.(50)8743-8761 doi:10.1039/d1cs00381j.
- Schulz, C. E., Castillo, R. G., Pantazis, D. A., DeBeer, S., Neese, F. (2021). Structure-Spectroscopy Correlations for Intermediate Q of Soluble Methane Monooxygenase: Insights from QM/MM Calculations. JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 143(17), 6560-6577. doi:10.1021/jacs.1c01180.
- Mathe, Z., McCubbin Stepanic, O., Peredkov, S., DeBeer, S. (2021). Phosphorus K beta X-ray emission spectroscopy detects non-covalent interactions of phosphate biomolecules in situ. Chemical Science. doi:10.1039/d1sc01266e.
- Cutsail III, G. E., Ross, M. O., Rosenzweig, A. C., DeBeer, S. (2021). Towards a unified understanding of the copper sites in particulate methane monooxygenase: an X-ray absorption spectroscopic investigation dagger. Chemical Science, (12), 6194-6209. doi:10.1039/d1sc00676b.
- Gomez Castillo, R., Hahn, A. W., van Kuiken, B. E., Henthorn, J. T., McGale, J., DeBeer, S. (2021). Probing Physical Oxidation State by Resonant X-ray Emission Spectroscopy: Applications to Iron Model Complexes and Nitrogenase. Angewandte Chemie, International Edition in English, (60), 10112-10121 doi:10.1002/anie.202015669.
- Rosenbach, H., Walla, E., Cutsail III, G. E., Birrell, J. A., Pascual-Ortiz, M., DeBeer, S., Fleig, U., Span, I. (2021). The Asp1 pyrophosphatase from S. pombe hosts a [2Fe-2S]2+ cluster in vivo. Journal of Biological Inorganic Chemistry. https://doi:10.1007/s00775-020-01840-w.
- Peters, J. W., Einsle, O., Dean, D. R., DeBeer, S., Hoffman, B. M., Holland, P. L., Seefeldt, L. C. (2021). Comment on "Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase". Science, 371(6530). https://doi:10.1126/science.abe5481.
- Duan, P.-C., Schulz, R. A., Römer, A., Van Kuiken, B. E., Dechert, S., Demeshko, S., Cutsail III, G. E., DeBeer, S., Mata, R. A., Meyer, F. (2021). Ligand Protonation Triggers H-2 Release from a Dinickel Dihydride Complex to Give a Doubly "T"-Shaped Dinickel(I) Metallodiradical. Angewandte Chemie, International Edition in English, 60(4), 1891-1896. https://doi:10.1002/anie.202011494.
- Rengshausen, S., Van Stappen, C., Levin, N., Tricard, S., Luska, K.L., DeBeer, S., Chaudret, B., Bordet, A., Leitner, W. (2021). Organometallic Synthesis of Bimetallic Cobalt‐Rhodium Nanoparticles in Supported Ionic Liquid Phases (CoxRh100−x@SILP) as Catalysts for the Selective Hydrogenation of Multifunctional Aromatic Substrates Small, 17, 2006683 (10pp) https://doi.org/10.1002/smll.202006683
- Van Stappen, C., Decamps, L., DeBeer, S. (2021). Preparation and Spectroscopic Characterization of Lyophilized Mo Nitrogenase Journal of the Biological Inorganic Chemistry. 26:81–91 https://doi.org/10.1007/s00775-020-01838-4
- Rodríguez-Maciá, P., Breuer, N., DeBeer, S., Birrell, J.A. (2020). Insight into the Redox Behavior of the [4Fe–4S] Subcluster in [FeFe] Hydrogenases ACS Catalysis 10(21), 13084-13095. https://doi.org/10.1021/acscatal.0c02771
- McCubbin Stepanic, O., Ward, J., Penner-Hahn, J.E., Deb, A., Bergmann, U., DeBeer, S. (2020). Probing a silent metal: A Combined X-ray Absorption and Emission Spectroscopic Study of Biologically Relevant Zinc Complexes Inorganic Chemistry. 59, 18, 13551–13560 https://doi.org/10.1021/acs.inorgchem.0c01931
- Jensen, K.M. Ø., DeBeer, S., Koziej, D. (2020). Editorial: Spectroscopy and scattering for chemistry: new possibilities and challenges with large scale facilities Nanoscale 12(35), 17968-17970. https://doi.org/10.1039/D0NR90182B
- Zimmermann, P., Peredkov, S., Abdala P.M., DeBeer, S., Tromp, M., Müller, C., van Bokhoven, J.A. (2020). Modern X-ray spectroscopy: XAS and XES in the laboratory Coordination Chemistry Reviews 423, 213466. https://doi.org/10.1016/j.ccr.2020.213466
- Budiyanto, E., Yu, M., Chen, M., DeBeer, S., Rüdiger, O., Tüysüz, H. (2020). Tailoring Morphology and Electronic Structure of Cobalt Iron Oxide Nanowires for Electrochemical Oxygen Evolution Reaction ACS Applied Energy Materials 3(9), 8583-8594. https://doi.org/10.1021/acsaem.0c01201
- Beheshti Askari, A., al Samarai, M., Hiraoka, N., Ishii, H., Tillmann, L., Muhler, M., DeBeer, S. (2020). In situ X-ray emission and high-resolution X-ray absorption spectroscopy applied to Ni-based Bimetallic Dry Methane Reforming Catalysts Nanoscale 20(28), 15185-15192. https://doi.org/10.1039/D0NR01960G
- Yu, M., Moon, G.-H., Castillo, R.G., DeBeer, S., Weidenthaler, C., Tüysüz, H. (2020). Dual Role of Silver Moieties Coupled with Ordered Mesoporous Cobalt Oxide towards Electrocatalytic Oxygen Evolution Reaction Angewandte Chemie International Edition 59(38), 16544-16552. https://doi.org/10.1002/anie.202003801
- DeBeer, S. (2020). Introduction to X-ray spectroscopy – including X-ray absorption, X-ray emission and resonant inelastic X-ray scattering Bioorganometallic Chemistry 407-432. https://doi.org/10.1515/9783110496574-011
- Rodríguez-Maciá, P., Galle, L., Bjornsson, R., Lorent, C., Zebger, I., Yoda, Y., Cramer, S., DeBeer, S., Span, I., Birrell, J.A. (2020). Caught in the Hinact: Crystal Structure and Spectroscopy Reveal a Sulfur Bound to the Active Site of an O2‐stable State of [FeFe] Hydrogenase Angewandte Chemie International Edition 59(38), 16786-16794. https://doi.org/10.1002/anie.202005208
- Levin, N., Peredkov, S., Weyhermüller, T., Rüdiger, O., Pereira, N.B., Grötzsch, D., Kalinko, A., DeBeer, S. (2020). Ruthenium 4d-to-2p X-ray Emission Spectroscopy: A Simultaneous Probe of the Metal and the Bound Ligands Inorganic Chemistry 59(12), 8272-8283. https://doi.org/10.1021/acs.inorgchem.0c00663
- Castillo, R.G., Henthorn, J.T., McGale, J., Maganas, D., DeBeer, S. (2020). Kβ X‐ray Emission Spectroscopic study of a second‐row transition metal (Mo) and its application to nitrogenase related model complexes Angewandte Chemie International Edition 59(31), 12965-12975. https://doi.org/10.1002/anie.202003621
- Beheshti-Askari. A., al Samarai, M., Morana, B., Tillmann, L., Pfänder, N., Wandzilak, A., Watts, B., Belkhou, R., Muhler, M., DeBeer, S. (2020). In-situ X-ray Microscopy reveals particle dynamics in a NiCo dry methane reforming catalyst under operating conditions ACS Catalysis 10(11), 6223-6230. https://doi.org/10.1021/acscatal.9b05517
- Van Stappen, C., Decamps, L., Cutsail III, G.E., Bjornsson, R., Henthorn, J.T., Birrell, J.A., DeBeer, S. (2020). The Spectroscopy of Nitrogenases Chemical Reviews 120(12), 5005-5081. https://doi.org/10.1021/acs.chemrev.9b00650
- Spiller, N., Chilkuri, V.G., DeBeer, S., Neese, F. (2020). Sulfur vs. Selenium as Bridging Ligand in Di‐Iron Complexes: A Theoretical Analysis European Journal of Inorganic Chemistry 2020(15-16), 1525-1538. https://doi.org/10.1002/ejic.202000033
- Maganas, D., Kowalska, J.K., Van Stappen, C., DeBeer, S., Neese, F. (2020). Mechanism of L2,3-edge X-Ray Magnetic Circular Dichroism Intensity from Quantum Chemical Calculations and Experiment - A case study on V(IV)/V(III) complexes The Journal of Chemical Physics 152(11), 114107. https://doi.org/10.1063/1.5129029
- Cutsail III, G.E., Blaesi, E.J., Pollock, C.J., Bollinger Jr, J.M., Krebs, C., DeBeer, S. (2020). High-resolution iron X-ray absorption spectroscopic and computational studies of non-heme diiron peroxo intermediates Journal of Inorganic Biochemistry 203, 110877. https://doi.org/10.1016/j.jinorgbio.2019.110877
- Birrell, J.A., Pelmenschikov, V., Mishra, N., Wang, H., Yoda, Y., Tamasaku, K., Rauchfuss, T.B., Cramer, S.P., Lubitz, W., DeBeer, S. (2020). Spectroscopic and Computational Evidence that [FeFe] Hydrogenases Operate Exclusively with CO-bridged Intermediates Journal of the American Chemical Society 142(1), 222-232. https://doi.org/10.1021/jacs.9b09745
- Chilkuri, V.G., DeBeer, S., Neese, F. (2020). Ligand Field Theory and Angular Overlap Model Based Analysis of the Electronic Structure of Homovalent Iron–Sulfur Dimers Inorganic Chemistry 59(2), 984-995. https://doi.org/10.1021/acs.inorgchem.9b00974
- Liu, Y., Resch, S.G., Klawitter, I., Cutsail III, G.E., Demeshko, S., Dechert, S., Kühn, F.E., DeBeer, S., Meyer, F. (2020). An Adaptable N‐Heterocyclic Carbene Macrocycle Hosting Copper in three Oxidation States Angewandte Chemie International Edition 59(14), 5696-5705. https://doi.org/10.1002/anie.201912745
- Mathe, Z., Pantazis, D.A., Lee, H.B., Gnewkow, R., Van Kuiken, B., Agapie, T., DeBeer, S. (2019). Calcium Valence-to-Core X-ray Emission Spectroscopy: A Sensitive Probe of Oxo Protonation in Structural Models of the Oxygen-Evolving Complex Inorganic Chemistry 58(23), 16292-16301. https://doi.org/10.1021/acs.inorgchem.9b02866
- DeRosha, D.E., Chilkuri, V.G., Van Stappen, C., Bill, E., Mercado, B.Q., DeBeer, S., Neese, F., Holland, P.L. (2019). Planar three-coordinate iron sulfide in a synthetic [4Fe-3S] cluster with biomimetic reactivity Nature Chemistry 11, 1019–1025. https://doi.org/10.1038/s41557-019-0341-7
- McGale, J., Cutsail, G.E. III, Joseph, C., Rose, M.J., DeBeer, S. (2019). Spectroscopic X-ray and Mössbauer Characterization of M6 and M5 Iron(Molybdenum)-Carbonyl Carbide Clusters: High Carbide-Iron Covalency Enhances Local Iron Site Electron Density Despite Cluster Oxidation Inorganic Chemistry 58(19), 12918-12932. https://doi.org/10.1021/acs.inorgchem.9b01870
- Al Samarai, M., Hahn, A.W., Askari, A.B., Cui, Y.-T., Yamazoe, K., Miyawaki, J., Harada, Y., Rüdiger, O., DeBeer, S. (2019). Elucidation of Structure-Activity Correlations in a Nickel-Manganese Oxide OER Catalyst by Operando Ni L-edge XAS and 2p3d RIXS ACS Applied Materials and Interfaces 11(42), 38595-38605. https://doi.org/10.1021/acsami.9b06752
- Van Stappen, C., Thorhallsson, A.T., Decamps, L., Bjornsson, R., DeBeer, S. (2019). Resolving the structure of the E1 state of Mo Nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations Chemical Science 10(42), 9807-9821. https://doi.org/10.1039/c9sc02187f
- Van Stappen, C., Davydov, R., Yang, Z.-Y., Fan, R., Guo, Y., Bill, E., Seefeldt, L.C., Hoffman, B.M., DeBeer, S. (2019). A spectroscopic description of the E1 state of Mo Nitrogenase based on Mo and Fe X-ray absorption and Mössbauer studies Inorganic Chemistry 58(18), 12365-12376. https://doi.org/10.1021/acs.inorgchem.9b01951
- Speelman, A.L., Čorić, I., Van Stappen, C., DeBeer, S., Mercado, B.Q., Holland, P.L. (2019). Nitrogenase-Relevant Reactivity of a Synthetic Iron–Sulfur–Carbon Site Journal of the American Chemical Society 141(33), 13148-13157. https://doi.org/10.1021/jacs.9b05353
- Chrysina, M., Heyno, E., Kutin Y., Reus, M., Nilsson, H., Nowaczyk. M.N., DeBeer, S., Neese, F., Messinger, J., Lubitz, W., Cox, N. (2019). Five-coordinate MnIV intermediate in the activation of nature’s water splitting cofactor Proceedings of the National Academy of Sciences of the United States of America 116(34), 16841-16846. https://doi.org/10.1073/pnas.1817526116
- Henthorn, J.T., Arias, R.J., Koroidov, S., Kroll, T., Sokaras, D., Bergmann, U., Rees, D.C., DeBeer, S. (2019). Localized Electronic Structure of Nitrogenase FeMoco Revealed by Selenium K-edge High Resolution X-ray Absorption Spectroscopy Journal of the American Chemical Society 141(34), 13676-13688. https://doi.org/10.1021/jacs.9b06988
- Yogendra, S., Weyhermüller, T., Hahn, A.W., DeBeer, S. (2019). From Ylides to Doubly Yldiide-Bridged Iron(II) High Spin Dimers via Self-Protolysis Inorganic Chemistry 58(14), 9358-9367. https://doi.org/10.1021/acs.inorgchem.9b01086
- Kowalska, J.K., Henthorn, J.T., Van Stappen, C., Trncik, C., Einsle, O., Keavney, D., DeBeer, S. (2019). X-ray Magnetic Circular Dichroism Spectroscopy Applied to Nitrogenase and Related Models: Experimental Evidence for a Spin-Coupled Mo(III) Angewandte Chemie International Edition 58(28), 9373-9377. https://doi.org/10.1002/anie.201901899
- Cutsail III, G.E., Gagnon, N.L., Spaeth, A.D., Tolman, W.B., DeBeer, S. (2019). Valence‐to‐Core X‐ray Emission Spectroscopy as a Probe of O‐O Bond Activation in Cu2O2 complexes Angewandte Chemie International Edition 58(27), 9114-9119. https://doi.org/10.1002/anie.201903749
- Kalläne, S.I., Hahn, A.W., Weyhermüller, T., Bill, E., Neese, F., DeBeer, S., van Gastel, M. (2019). Spectroscopic and Quantum Chemical Investigation of Benzene-1,2- dithiolate-Coordinated Diiron Complexes with Relevance to Dinitrogen Activation Inorganic Chemistry 58(8), 5111-5125. https://doi.org/10.1021/acs.inorgchem.9b00177
- Maganas, D., Kowalska, J.K., Noiijen, M., DeBeer, S., Neese, F. (2019). Comparison of Multireference Ab initio Wavefunction Methodologies for X-Ray Absorption Edges: A Case study on [Fe(II/III)Cl4]2-/1- molecules The Journal of Chemical Physics 150(10), 104106. https://doi.org/10.1063/1.5051613
- Malzer, W., Grötzsch, D., Gnewkow, R., Schlesiger, C., Kowalewski, F., Van Kuiken, B., DeBeer, S., Kanngießer, B. (2018). A laboratory spectrometer for high throughput X-ray emission spectroscopy in catalysis research Review of Scientific Instruments 89, 113111. https://doi.org/10.1063/1.5035171
- Cutsail III, G.E., Banerjee, R., Zhou, A., Que, L., Lipscomb, J.D., DeBeer, S. (2018). High-Resolution EXAFS Provides Evidence for a Longer Fe•••Fe Distance in the Q Intermediate of Methane Monooxygenase Journal of American Chemical Society 140(48) 16807-16820. https://doi.org/10.1021/jacs.8b10313
- Hahn, A.W., Van Kuiken, B.E., Chilkuri, V.G., Levin, N., Bill, E., Weyhermüller, T., Nicolaou, A., Miyawaki, J., Harada, Y., DeBeer, S. (2018). Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X ray Scattering (RIXS) Inorganic Chemistry 57(37), 11918-11923. https://doi.org/10.1021/acs.inorgchem.8b01550
- Rodriguez-Maciá, P., Reijerse, E.J., van Gastel, M., DeBeer, S., Lubitz, W., Rüdiger, O., Birrell, J.A. (2018). Sulfide Protects [FeFe] Hydrogenases From O2 Journal of the American Chemical Society 140(30), 9346-9350. https://doi.org/10.1021/jacs.8b04339
- Chantzis, A., Kowalska, J.K., Maganas, D., DeBeer, S., Neese, F. (2018). Ab initio Wavefunction-based Determination of Element Specific Shifts for the Efficient Calculation of X-Ray Absorption Spectra of Main Group Elements and First Row Transition Metals Journal of Chemical Theory and Computation 14(7), 3686-3702. https://doi.org/10.1021/acs.jctc.8b00249
- Galle, L.M., Cutsail, G.E. III, Nischwitz, V., DeBeer, S., Span, I. (2018). Spectroscopic characterization of the Co-substituted C-terminal domain of rubredoxin-2 Biological Chemistry 399(7), 787-798. https://doi.org/10.1515/hsz-2018-0142
- Van Kuiken, B.E., Hahn, A.W., Nayyar, B., Schiewer, C.E., Lee, S.C., Meyer, F., Weyhermüller, T., Nicolaou, A., Cui, Y-T., Miyawaki, J., Hatada, Y., DeBeer, S. (2018). Electronic Spectra of Iron-Sulfur Complexes Measured by 2p3d RIXS Spectroscopy Inorganic Chemistry 57(12), 7355-7361. https://doi.org/10.1021/acs.inorgchem.8b01010
- Van Stappen, C., Maganas, D., DeBeer, S., Bill, E., Neese, F. (2018). Investigation of the Magnetic and Spectroscopic Properties of V(III) and V(IV) Complexes Inorganic Chemistry 57(11), 6421-6438. https://doi.org/10.1021/acs.inorgchem.8b00486
- Maganas, D., DeBeer, S., Neese, F. (2018).A Pair Natural Orbitals Restricted Open Shell Configuration Interaction (PNO-ROCIS) Approach for Calculating X-ray Absorption Spectra of Large Chemical Systems Journal of Physical Chemistry A 122(5), 1215-1227. https://doi.org/10.1021/acs.jpca.7b10880
- Leipzig, B.K., Rees, J.A., Kowalska, J.K., Theisen, R.M., Kavčič, M., Poon, P.C.Y., Kaminsky, W., DeBeer, S., Bill, E., Kovacs, J.A. (2018). How Do Ring Size and π-Donating Thiolate Ligands Affect Redox-Active, α-Imino-N-heterocycle Ligand Activation? Inorganic Chemistry 57(4), 1935-1949. https://doi.org/10.1021/acs.inorgchem.7b02748
- DeBeer, S. (2018). Advanced X-ray Spectroscopic Methods for Studying Iron-Sulfur-Containing Proteins and Model Complexes Methods Enzymology. Fe-S Cluster Enzymes Part B 599, 427-450. https://doi.org/10.1016/bs.mie.2017.09.008
- Römelt, C., Song, J.S., Tarrago, M., Rees, J.A., van Gastel, M., Weyhermüller, T., DeBeer, S., Bill, E., Neese, F., Ye, S. (2017). Electronic Structure of a Formal Iron(0), Porphyrin Complex Relevant to CO2 Reduction Inorganic Chemistry 56(8), 4745-4750. https://doi.org/10.1021/acs.inorgchem.7b00401
- Rees, J.A., Bjornsson, R., Kowalska, J.K., Lima, F.A., Schlesier, J., Sippel, D., Weyhermüller, T., Einsle, O., Kovacs, J.A., DeBeer, S. (2017). Comparative electronic structures of nitrogenase FeMoco and FeVco Dalton Transactions 46(8), 2445-2455. https://doi.org/10.1039/c7dt00128b
- Bjornsson, R., Neese, F., DeBeer, S. (2017). Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge Inorganic Chemistry 56(3), 1470-1477. https://doi.org/10.1021/acs.inorgchem.6b02540
- Castillo, R.G., Banerjee, R., Allpress, C.J., Rohde, G.T., Bill, E., Que Jr., L., Lipscomb, J.D., DeBeer, S. (2017). High-Energy-Resolution Fluorescence-Detected X-ray Absorption of the Q Intermediate of Soluble Methane Monooxygenase Journal of the American Chemical Society 139(49), 18024-18033. https://doi.org/10.1021/jacs.7b09560
- Maganas, D., DeBeer, S., Neese, F. (2017). A Restricted Open Configuration Interaction with Singles Method To Calculate Valence-to-Core Resonant X-ray Emission Spectra: A Case Study Inorganic Chemistry 56(19), 11819-11836. https://doi.org/10.1021/acs.inorgchem.7b01810
- Koziej, D., DeBeer, S. (2017). Application of Modern X-ray Spectroscopy in Chemistry-Beyond Studying the Oxidation State Chemistry of Materials 29(17), 7051-7053. https://doi.org/10.1021/acs.chemmater.7b03455
- Chilkuri, V.G., DeBeer, S., Neese, F. (2017). Revisiting the Electronic Structure of FeS Monomers Using ab Initio Ligand Field Theory and the Angular Overlap Model Inorganic Chemistry 56(17), 10418-10436. https://doi.org/10.1021/acs.inorgchem.7b01371
- Kowalska, J.K., Nayyar, B., Rees, J.A., Schiewer, C.E., Lee, S.C., Kovacs, J.A., Meyer, F., Weyhermüller, T., Otero, E., DeBeer, S. (2017). Iron L2,3-edge X-ray Absorption and X-ray Magnetic Circular Dichroism Studies of Molecular Iron Complexes with Relevance to the FeMoco and FeVco Active Sites of Nitrogenase Inorganic Chemistry 56(14), 8147-8158. https://doi.org/10.1021/acs.inorgchem.7b00852
- Hahn, A.W., Van Kuiken, B.E., al Samarai, M., Atanasov, M., Weyhermüller, T., Cui, Y.T., Miyawaki, J., Harada, Y., Nicolaou, A., DeBeer, S. (2017). Measurement of the Ligand Field Spectra of Ferrous and Ferric Iron Chlorides Using 2p3d RIXS Inorganic Chemistry 56(14), 8203-8211. https://doi.org/10.1021/acs.inorgchem.7b00940
- Casitas, A., Rees, J.A., Goddard, R., Bill, E., DeBeer, S., Fürstner, A. (2017). Two Exceptional Homoleptic Iron(IV) Tetraalkyl Complexes Angewandte Chemie International Edition 56(34), 10108-10113. https://doi.org/10.1002/anie.201612299
- Van Kuiken, B.E., Hahn, A.W., Maganas, D., DeBeer, S. (2016). Measuring Spin-Allowed and Spin-Forbidden d-d Excitations in Vanadium Complexes with 2p3d Resonant Inelastic X-ray Scattering Inorganic Chemistry 55(21), 11497-11501. https://doi.org/10.1021/acs.inorgchem.6b02053
- Kowalska, J.K., Lima, F.A., Pollock, C. J., Rees, J.A., DeBeer, S. (2016). A Practical Guide to High-resolution X-ray Spectroscopic Measurements and their Applications in Bioinorganic Chemistry Israel Journal of Chemistry 56(9-10), 803-815. https://doi.org/10.1002/ijch.201600037
- Rees, J.A., Wandzilak, A., Maganas, D., Wurster, N.I.C., Hugenbruch, S., Kowalska, J.K., Pollock, C.J., Lima, F.A., Finkelstein, K.D., DeBeer, S. (2016). Experimental and theoretical correlations between vanadium K-edge X-ray absorption and Kß emission spectra Journal of Biological Inorganic Chemistry 21(5-6), 793-805. https://doi.org/10.1007/s00775-016-1358-7
- Kupper, C., Rees, J.A., Dechert, S., DeBeer, S., Meyer, F. (2016). Complete Series of {FeNO}8, {FeNO}7, and {FeNO}6 Complexes Stabilized by a Tetracarbene Macrocycle Journal of the American Chemical Society 138(25), 7888-7898. https://doi.org/10.1021/jacs.6b00584
- Kowalska, J.K., Hahn, A.W., Albers, A., Schiewer, C.E., Bjornsson, R., Lima, F.A., Meyer, F., DeBeer, S. (2016). X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2]n Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron-Sulfur Clusters Inorganic Chemistry 55(9), 4485-4497. https://doi.org/10.1021/acs.inorgchem.6b00295
- Hugenbruch, S., Shafaat, H.S., Krämer, T., Delgado-Jaime, M.U., Weber, K., Neese, F., Lubitz, W., DeBeer, S. (2016). In search of metal hydrides: an X-ray absorption and emission study of [NiFe] hydrogenase model complexes Physical Chemistry Chemical Physics 18(16), 10688-10699. https://doi.org/10.1039/c5cp07293j
- Martin-Diaconescu, V., Chacon, K.N., Delgado-Jaime, M.U., Sokaras, D., Weng, T.C., DeBeer, S., Blackburn, N.J. (2016). Kß Valence to Core X-ray Emission Studies of Cu(I) Binding Proteins with Mixed Methionine - Histidine Coordination. Relevance to the Reactivity of the M- and H-sites of Peptidylglycine Monooxygenase Inorganic Chemistry 55(7), 3431-3439. https://doi.org/10.1021/acs.inorgchem.5b02842
- DeBeer, S., Bergmann U. (2016). X-ray Emission Spectroscopic Techniques in Bioinorganic Applications Encyclopedia of Inorganic and Bioinorganic Chemistry 1-14. https://doi.org/10.1002/9781119951438.eibc2158
Preise
- 2023 Glenn T. Seaborg Lectureship, University of California Berkeley
- 2022 R.J.P. Williams Lectureship, Oxford University
- 2022 Malcom H. Chisholm Lecturer, The Ohio State University
- 2021 Fellow of the Royal Society of Chemistry
- 2019 European Research Council Synergy Grant Awardee
- 2016 Inorganic Chemistry Lectureship Award
- 2015 Society of Biological Inorganic Chemistry, Early Career Award
- 2013 European Research Council Consolidator Grant Awardee
- 2012 Kavli Fellow, U.S. National Academy of Sciences
- 2011-2013 Alfred P. Sloan Research Fellow
Abteilungsmitglieder
Sekretariat
Valerie EstersGruppenleiter*innen
Dr. Sergey PeredkovDr. Christina Römelt
Dr. Olaf Rüdiger
Dr. Kushal Sengupta
Dr. Thomas Weyhermüller
Projektleiter*innen
Dr. Yves KayserDr. Sergio Augusto Venturinelli Jannuzzi
Gruppen





Projekte


Anorganische Spektroskopie

Unsere Abteilung ist auf die Entwicklung und Anwendung fortschrittlicher röntgenspektroskopischer Methoden fokussiert, um Vorgänge in der chemischen und biologischen Katalyse zu verstehen. Wir sind an der Entwicklung von statischen und zeitaufgelösten Verfahren zur Erforschung der elektronischen Struktur von Übergangsmetallverbindungen interessiert, aber auch an der Entwicklung von zweidimensionalen Verfahren, die eine erhöhte Selektivität ermöglichen. Angetrieben werden wir dabei durch drängende Fragen der Energieforschung. Wie können etwa starke chemische Bindungen durch leicht verfügbare Metalle, wie z.B. Eisen und Kupfer, aktiviert werden? In diesem Zusammenhang konzentrieren wir uns auf vier Metall-katalysierte Reaktionen:
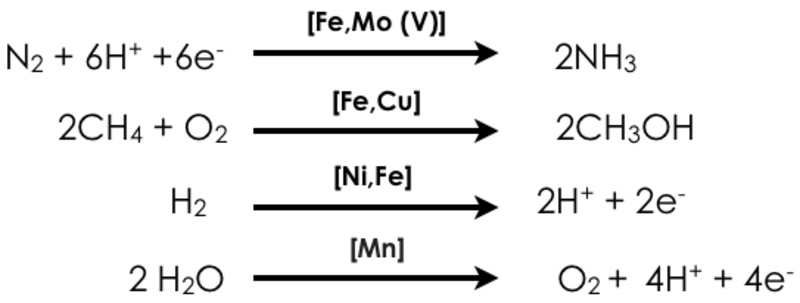

In den oben dargestellten Reaktionen sind sowohl biologische (homogene) als auch heterogene Katalysatoren vertreten, die die Umsetzungen mit unterschiedlicher Effizienz und manchmal in mehreren Schritten vorantreiben (z.B. bei der industriellen Methan Oxidation). Wir sind daran interessiert, die elektronische Struktur der Katalysatoren und die Natur der Intermediate im Reaktionszyklus zu verstehen. Im Idealfall ist das so gewonnene Wissen die Basis zur Entwicklung neuer, verbesserter Katalysatoren. Unser Ansatz nutzt röntgenbasierte spektroskopische Methoden, die sowohl resonante (RXES, RIXS), als auch nicht resonante (XES) Röntgenemissions-, oder Röntgenabsorptions-Vorgänge (XAS) analysieren. Zusätzlich werden Röntgen-Circulardichroismus (XMCD), und kernresonante Schwingungsspektroskopie (NRVS) genutzt (Abb. 1). Neben unseren Synchrotron-basierten Aktivitäten entwickeln wir auch Instrumente zur hausinternen Röntgenspektroskopie. Die Kombination der verschiedenen Methoden ermöglicht es, Informationen über den Oxidationszustand, den Spinzustand und die lokale chemische Umgebung des katalytisch aktiven Metallzentrums zu gewinnen. Unsere Studien werden durch weitere Messungen wie UV-vis, EPR, Raman und Mössbauerspektroskopie sowie durch moderne quantenchemische Rechnungen ergänzt, um elektronische und strukturelle Änderungen des katalytischen Systems zu untersuchen.




1. Methodenentwicklung
1.1. Valenz XES. In der Vergangenheit hat sich unsere Arbeitsgruppe darauf konzentriert, den vollständigen Informationsgehalt der Röntgenemissionen von Valenzübergängen (XES) zur Untersuchung von biologischen Katalysatoren nutzbar zu machen.1-12 Bei einer Valenz XES Messung werden zunächst 1s Elektronen am Metall herausgeschlagen und die daraufhin folgenden Fluoreszenzemissionen aufgezeichnet, die beim Auffüllen der 1s Orbitale mit Valenzelektronen auftreten. Auf diese Weise liefern Valenz XES Spektren ein Abbild der Ionisationspotentiale der gebundenen Liganden, die eine starke Abhängigkeit von der Art des Elements (z.B. C, N, O) und des Portionierungszustandes (O2-, OH-, H2O) liefern (Abb. 2).
Ein bemerkenswertes Beispiel für die erfolgreiche Anwendung der Methode ist etwa die Identifizierung eines zentralen Kohlenstoffatoms im FeMoco Cluster der Nitrogenase.8 Dieses zentrale Atom hatte sich jahrzehntelang einer Identifizierung durch andere Verfahren entzogen und verdeutlicht somit die Leistungsfähigkeit der XES. Neuere Studien der Gruppe Anorganische Spektroskopie beschäftigen sich mit der Nutzung der XES zur Aufklärung der Biosynthese von Metallokofaktoren und der atomaren Zusammensetzung des FeVco in Vanadium-Nitrogenase.7,13,14
Valenz XES ist nicht nur empfindlich bezüglich der Identität eines Liganden, sondern auch in welchem Maße dieser aktiviert ist (Schwächung der Bindung). Beispielsweise konnten in XES Spektren von Fe-N2 Komplexen bestimmte Merkmale festgestellt werden, die den Bereich des Spektrums betreffen, der mit der Interaktion des N2 2s2s s* Orbitals mit dem Eisenzentrum korreliert ist. Wird die N-N Bindung beim Übergang von der N-N Dreifachbindung zum Nitrid hin länger, nimmt die Energie des Übergangs um etwa 2 eV ab. Dies zeigt, dass die Fe Valenz XES eine ideale spektrale Sonde zur Untersuchung der N-N Bindungsspaltung (oder die Spaltung anderer zweiatomiger Liganden) am Eisen (oder anderer Metalle) ist.
Zu diesem Zweck entwickeln wir die dispersive zeitaufgelöste Valenz XES zum Studium der Übergangsmetallkatalyse. Kürzlich konnte, durch Förderung mittels eines ERC Consolidator Grant (N2RED, PI: DeBeer), ein in-house Setup installiert werden, das in Zusammenarbeit mit Prof. Kanngießer an der TU Berlin entwickelt wurde. Das Instrument nutzt eine Labor-Röntgenquelle (Excillium, Metal Jet) in Kombination mit einem zylindrischen pyrolytischen Graphit Kristall (HAPG) und einem CCD Detektor um XES im Bereich 2.4-9 keV zu messen (Abb. 3).
Dieses einzigartige in-house Instrument reduziert den Bedarf für Messzeit am Synchrotron, da es ideale Möglichkeiten bietet, konzentrierte Proben zu untersuchen und aufwendigere Messungen am Synchrotron vorzubereiten. Im Bereich der dispersiven Synchrotron-basierten XES leitet unsere Abteilung auch die Entwicklung der PINK Beam Line am Energy Materials In-situ Laboratory in Berlin. PINK eröffnet uns auch die Möglichkeit an heterometallischen Katalysatoren gleichzeitig XES Messungen bei zwei verschiedenen Wellenlängen vorzunehmen. Diese Arbeiten werden von Dr. Sergey Peredkov geleitet.
1.2. Resonante Valenz XES. Um die Selektivität der XES weiter zu verbessern, entwickeln wir die resonante Valenz XES.2 Dies ist ein zweidimensionales Verfahren bei dem man resonant ein 1s Elektron in teilweise besetzte oder unbesetzte Zustände des Absorber-Atoms anregt (das Äquivalent eines Scans der Röntgenabsorptionskante) während gleichzeitig die energetisch am höchsten liegenden Emissionsvorgänge aufgezeichnet werden (die Valenz XES). Dadurch ist man in der Lage, eine zweidimensionale Darstellung zu konstruieren, in der die einfallende Energie auf der X-Achse und die emittierte Energie auf der Y-Achse aufgetragen ist (Abb. 4). Die horizontalen Schnitte durch diese Ebene repräsentieren dann die XAS Daten bei einer gegebenen Emissionsenergie und können auch als "ligandenselektive XAS“ bezeichnet werden. Die vertikalen Schnitte zeigen dann die XES Daten bei einer gegebenen einfallenden Energie (Valenz RXES). Wir glauben, dass dieser Ansatz ein hervorragendes Mittel darstellt um XAS Daten verschiedener Metall-Ligand Wechselwirkungen in komplexen Materialen zu messen.
1.3. 2p3d RIXS. Zusätzlich zur Valenz REXS entwickeln wir Verfahren, um den vollständigen Informationsgehalt von Übergangsmetall 2p3d RXES, oft auch 2p3d RIXS genannt, zu erschließen.15
2p3d RIXS ist mit dem Dipol-erlaubten Übergang des 2p63dn Grundzustandes in den 2p53dn+1 Zwischenzustand verknüpft, der dann wieder, Dipol-erlaubt, in einen finalen 2p63dn Zustand relaxiert (Abb. 5). Somit entspricht der Übergang zwischen dem Grundzustand und dem finalen Zustand formal einem Dipol-verbotenen d-d Übergang zu dem man durch zwei Dipol-erlaubte Vorgänge gelangt. In einer 2p3d RIXS Messung bezeichnet man die Energiedifferenz zwischen dem Grundzustand und dem finalen Zustand als “Energy Tranfer“ Achse, welche, der Natur des Experiments entsprechend, zwischen 0-10 eV (oder 0 bis über 80.000 cm-1) angesiedelt ist. Dies bedeutet, dass die spektralen Bereiche, die sonst nicht durch optische oder infrarote Standard-Messungen zugänglich sind, hier erreichbar sind. Zudem konnte in bestimmten Fällen durch 2p3d RIXS Experimente gezeigt werden, dass Spin-verbotene Übergänge wegen der Spin-Bahn-Kopplung zum Zwischenzustand beobachtet werden können. Somit sollte es möglich sein, gewissermaßen die komplette Spin-Leiter zu vermessen und damit nie dagewesene experimentelle Einblicke in die elektronische Struktur von Übergangsmetallverbindungen zu erlangen.
2. Anwendungen
Die im vorigen Abschnitt diskutierten neu entwickelten spektroskopischen Methoden bilden eine gute Basis für Anwendungen in der biologischen und chemischen Katalyseforschung. Im Folgenden stellen wir kurz einige unserer gegenwärtigen und geplanten Forschungsthemen vor.
N2 Reduktion. Die Umwandlung von N2 in Ammoniak (NH3) ist von besonderer Bedeutung für das Leben auf der Erde. Dieser kinetisch ausgesprochen schwierige Prozess wird auf Seiten der Biologie durch das Enzym Nitrogenase bewerkstelligt und industriell mit hoher Effizienz durch den Haber-Bosch Prozess abgebildet. Chemiker haben dagegen bisher Schwierigkeiten einen homogenen Katalysator zu entwickeln der mit dem biologischen oder dem industriellen Prozess konkurrieren kann. Wir gehen davon aus, dass dieses bisherige “Unvermögen“ mit einem Mangel an Grundlagenwissen zu dieser Thematik zusammenhängt. Wie kann man die Spaltung der stärksten in der Chemie bekannten homodiatomaren Bindung effektiv bewerkstelligen? Wie wechselwirkt der metallhaltige Katalysator mit N2? Welche Intermediate werden beim Bindungsbruch des N2 Moleküls gebildet? Um diese Fragen für biologische und heterogene Katalysatoren zu beantworten, wenden wir fortschrittliche röntgenspektroskopische Methoden an. Für den biologischen Katalysator versuchen wir die komplexe elektronische Struktur des Grundzustandes, sowie die Rolle des zentralen Kohlenstoffatoms und die Funktion des Hetreometalls (Mo oder V) zu verstehen, darüber hinaus versuchen wir die Bindungsstelle des Substrats zu identifizieren. Beim heterogenen Katalysator versuchen wir die Rolle der an der Oberfläche gebildeten Eisenitrid-Funktionen, die Oxidationsstufe des aktiven Eisenzentrums und die Funktion des Kalium-Promoters zu bestimmen. Diese Fragestellungen werden hoffentlich durch die im vorhergehenden Abschnitt diskutierten fortschrittlichen spektroskopischen Methoden beantwortet.
CH4 Oxidation. Die biologische Oxidation von Methan zu Methanol durch lösliche Methanmonoxygenase ist ein Vorgang der im Zusammenhang einer Energiewirtschaft mit erneuerbaren Energien ausgesprochen interessant ist. Zurzeit forschen wir in der Gruppe Anorganische Spektroskopie intensiv daran, den Mechanismus der biologischen Methanoxidation aufzuklären und dabei vor allem die Struktur des kontrovers diskutierten Intermediates MMO-Q, welches für die Abstraktion eines Protons vom Methan zuständig ist, zu bestimmen. Durch die Anwendung von hochaufgelösten XAS Methoden konnten wir bereits neue Einblicke in die elektronische Struktur von MMO-Q gewinnen. Dies spornt uns zu weiteren “freeze-quench“ und in-situ Studien zur biologischen Methanoxidation an.
H2O Oxidation. Ähnlich wie im Fall der N2-Reduktion, sind wir sehr daran interessiert, die Mn-katalysierte Wasseroxidation sowohl im biologischen System (PSII), als auch in der heterogenen Katalyse zu verstehen. Wir nutzen auch hier unsere Methoden um Mn-Birnessite, oberflächendotierte Mn-Systeme und das aktive Zentrum des Photosystems II (Mn4Ca) zu untersuchen. Gleichzeitige und zeitaufgelöste XES an Mn- und Ca-Zentren bei zwei verschiedenen Energien wird dazu beitragen die Rolle der beiden Elemente bei der Katalyse zu entschlüsseln. Unser Ziel ist es die Faktoren die für eine O-O Bindungsbildung ausschlaggebend sind auf atomarer Grundlage zu verstehen.
H2 Produktion. Die Produktion und Aktivierung von Wasserstoff stand in den letzten Jahrzehnten im Fokus vieler Forschungsarbeiten, besonders auch in Bezug auf die Nutzung erneuerbarer Energien und von klimaneutralen Kraftstoffen. Die am weitesten verbreiteten und stabilsten Hydrogenasen enthalten im aktiven Zentrum eine [NiFe]-Einheit. Man nimmt an, dass das redox-aktive Nickel-Zentrum eine entscheidende Rolle bei der Bindung und Aktivierung von Wasserstoff spielt und viele Studien nutzten Röntgenstrukturanalyse, Elektrochemie, FTIR und EPR-Spektroskopie um die Aktivierung und den Mechanismus der Hydrogenase zu erforschen. Es wurden drei katalytisch relevante Intermediate gefunden von denen nur der sogenannte Ni-C Zustand paramagnetisch ist. Das aerob inaktivierte hydroxo-verbrückte Ni-B Intermediat und das hydrido-verbrückte Ni-C Intermediat wurden bisher als einzige Zustände strukturell gut charakterisiert. Darüber hinaus, wurde mit Hilfe von FTIR und Röntgenabsorptionsspektroskopie versucht, die beiden anderen Zustände, Ni-SI und Ni-R, zu charakterisieren. Allerdings bleiben nach wie vor viele Fragen offen, was beispielsweise die Nickel Spin-Zustände, die Natur des verbrückenden Liganden zwischen den Metallzentren im jeweiligen Redox-Zustand betrifft, oder etwa in welcher Art und an welches Metall der Wasserstoff bindet. Wir werden versuchen all diese Fragen durch eine Kombination unserer röntgenbasierten Methoden zu beantworten um schließlich den Mechanismus der biologischen Wasserstoff-Produktion zu entschlüsseln.
Referenzen
[1] Delgado-Jaime, M. U., Dible, B. R., Chiang, K. P., Brennessel, W. W., Bergmann, U., Holland, P. L., and DeBeer, S. (2011) Identification of a Single Light Atom within a Multinuclear Metal Cluster Using Valence-to-Core X-ray Emission Spectroscopy, Inorg Chem 50, 10709-10717.
[2] Hall, E. R., Pollock, C. J., Bendix, J., Collins, T. J., Glatzel, P., and DeBeer, S. (2014) Valence-to-Core-Detected X-ray Absorption Spectroscopy: Targeting Ligand Selectivity, J Am Chem Soc 136, 10076-10084.
[3] Hugenbruch, S., Shafaat, H. S., Kramer, T., Delgado-Jaime, M. U., Weber, K., Neese, F., Lubitz, W., and DeBeer, S. (2016) In search of metal hydrides: an X-ray absorption and emission study of [NiFe] hydrogenase model complexes, Phys Chem Chem Phys 18, 10688-10699.
[4] Kowalska, J., and DeBeer, S. (2015) The role of X-ray spectroscopy in understanding the geometric and electronic structure of nitrogenase, Bba-Mol Cell Res 1853, 1406-1415.
[5] Kowalska, J. K., Hahn, A. W., Albers, A., Schiewer, C. E., Bjornsson, R., Lima, F. A., Meyer, F., and DeBeer, S. (2016) X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2](n) Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron-Sulfur Clusters, Inorg Chem 55, 4485-4497.
[6] Kowalska, J. K., Lima, F. A., Pollock, C. J., Rees, J. A., and DeBeer, S. (2016) A Practical Guide to High-resolution X-ray Spectroscopic Measurements and their Applications in Bioinorganic Chemistry, Isr J Chem 56, 803-815.
[7] Lancaster, K. M., Hu, Y. L., Bergmann, U., Ribbe, M. W., and DeBeer, S. (2013) X-ray Spectroscopic Observation of an Interstitial Carbide in NifEN-Bound FeMoco Precursor, J Am Chem Soc 135, 610-612.
[8] Lancaster, K. M., Roemelt, M., Ettenhuber, P., Hu, Y. L., Ribbe, M. W., Neese, F., Bergmann, U., and DeBeer, S. (2011) X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor, Science 334, 974-977.
[9] Lee, N., Petrenko, T., Bergmann, U., Neese, F., and DeBeer, S. (2010) Probing Valence Orbital Composition with Iron K beta X-ray Emission Spectroscopy, J Am Chem Soc 132, 9715-9727.
[10] Pollock, C. J., and DeBeer, S. (2011) Valence-to-Core X-ray Emission Spectroscopy: A Sensitive Probe of the Nature of a Bound Ligand, J Am Chem Soc 133, 5594-5601.
[11] Pollock, C. J., and DeBeer, S. (2015) Insights into the Geometric and Electronic Structure of Transition Metal Centers from Valence-to-Core X-ray Emission Spectroscopy, Accounts Chem Res 48, 2967-2975.
[12] Pollock, C. J., Grubel, K., Holland, P. L., and DeBeer, S. (2013) Experimentally Quantifying Small-Molecule Bond Activation Using Valence-to-Core X-ray Emission Spectroscopy, J Am Chem Soc 135, 11803-11808.
[13] Rees, J. A., Martin-Diaconescu, V., Kovacs, J. A., and DeBeer, S. (2015) X-ray Absorption and Emission Study of Dioxygen Activation by a Small-Molecule Manganese Complex, Inorg Chem 54, 6410-6422.
[14] Rees, J. A., Bjornsson, R., Kowalska, J. K., Lima, F. A., Schlesier, J., Sippel, D., Weyhermueller, T., Einsle, O., Kovacs, J. A., and DeBeer, S. (2017) Comparative electronic structures of nitrogenase FeMoco and FeVco, Dalton T 46, 2445-2455.
[15] Van Kuiken, B. E., Hahn, A. W., Maganas, D., and DeBeer, S. (2016) Measuring Spin-Allowed and Spin-Forbidden d-d Excitations in Vanadium Complexes with 2p3d Resonant Inelastic X-ray Scattering, Inorg Chem 55, 11497-11501.